Na = sodium; eGFR = estimated glomerular filtration rate, BSA = body surface area, Cr Serum = serum creatinine; Cr Urine = urine creatinine; h = hours; Na Urine = urinary sodium concentration
Predicted sodium output using this equation has a strong correlation with the measured sodium output (r = 0.91, p < 0.0001), facilitating early discernment of natriuretic resistance to a diuretic dose [26]. Spot urine sodium is limited as a complete diuretic resistance metric because it does not incorporate diuretic dose. Unlike diuretic efficacy which lacks a threshold value, a predicted sodium output <50–70 mmol/L predicts a positive sodium balance with twice daily IV loop diuretic and is associated with worse heart failure outcomes [26, 29, 30]. By identifying patients with natriuretic resistance within 1–2 h, clinicians can make rapid diuretic titrations to overcome diuretic resistance.
Prognostic Impact of Diuretic Resistance in Acute Heart Failure
The prognostic impact of diuretic resistance varies with the definition of diuretic resistance employed and retains the defining metric’s limitations. At the most extreme, diuretic resistance requiring mechanical volume removal denotes a poor prognosis [31]. In the absence of randomized trials comparing therapies for diuretic resistance, it is unknown whether it is the presence of diuretic resistance itself or the resultant decongestive therapies that confer prognostic value. Loop diuretics increase neurohumoral activation regardless of the volume state, dose, or diuretic response [32, 33]. Initial literature evaluating prognosis focused on the diuretic dose, with an assumption of diuretic resistance. An almost linear association between increasing mortality and increasing IV furosemide daily doses occurred in a multivariate analysis of the ESCAPE [34]. Subsequent analyses adjusted for other covariates to investigate whether high loop diuretic doses were simply a surrogate for severity of illness or a driver of poor outcomes. After applying propensity score matching to balance for illness severity, an analysis of the Acute Heart Failure Global Survey of Standard Treatment (ALARM-HF) registry found that the association between IV diuretic dose and mortality was no longer present [19]. The DOSE-AHF trial randomized patients to a high or low dose loop diuretic strategy, allowing insight into the relative impact of the beneficial (decongestive) effects of higher diuretic dose and the potential harmful effects (neurohumoral activation) [35]. The DOSE trial found no effect on 60-day death or rehospitalization between those randomized to high or low dose IV diuretics, although the prevalence of diuretic resistance was unknown [36]. Further analyses of the DOSE-AHF trial found a benefit in 60-day outcomes in patients randomized to a high-dose loop diuretic strategy, after adjusting for cumulative dose, but this effect was eliminated after adjusting for the resulting net urine output [35]. Change in renin-angiotensin-aldosterone system (RAAS) biomarkers during diuresis did not differ between the high and low dose groups and were not associated with 60-day outcomes in the DOSE-AHF trial [33]. The potential for dose-related harm from loop diuretics cannot be excluded, although the decongestive benefits of high dose loop diuretics appear to offset potential adverse effects.
Diuretic efficiency, as a proxy of diuretic resistance, has been employed to separate the prognostic effect of decongestive therapy intensity from resistance itself. Using diuretic efficiency above and below the median value, patients in the ESCAPE with low diuretic efficiency had increased mortality (HR 3.57; 95% CI 1.46–8.73; p = 0.005), with those exhibiting low diuretic efficiency on high loop diuretic doses having the worst prognosis [20]. Consequently, diuretic resistance is known to confer a worse prognosis when: (1) high dose loop diuretics are required with sustained low diuretic efficiency or (2) resistance prohibits achievement of euvolemia with medical therapy.
Mechanisms of Loop Diuretic Resistance and Mechanism-Based Therapies to Restore Diuretic Efficacy in Acute Heart Failure
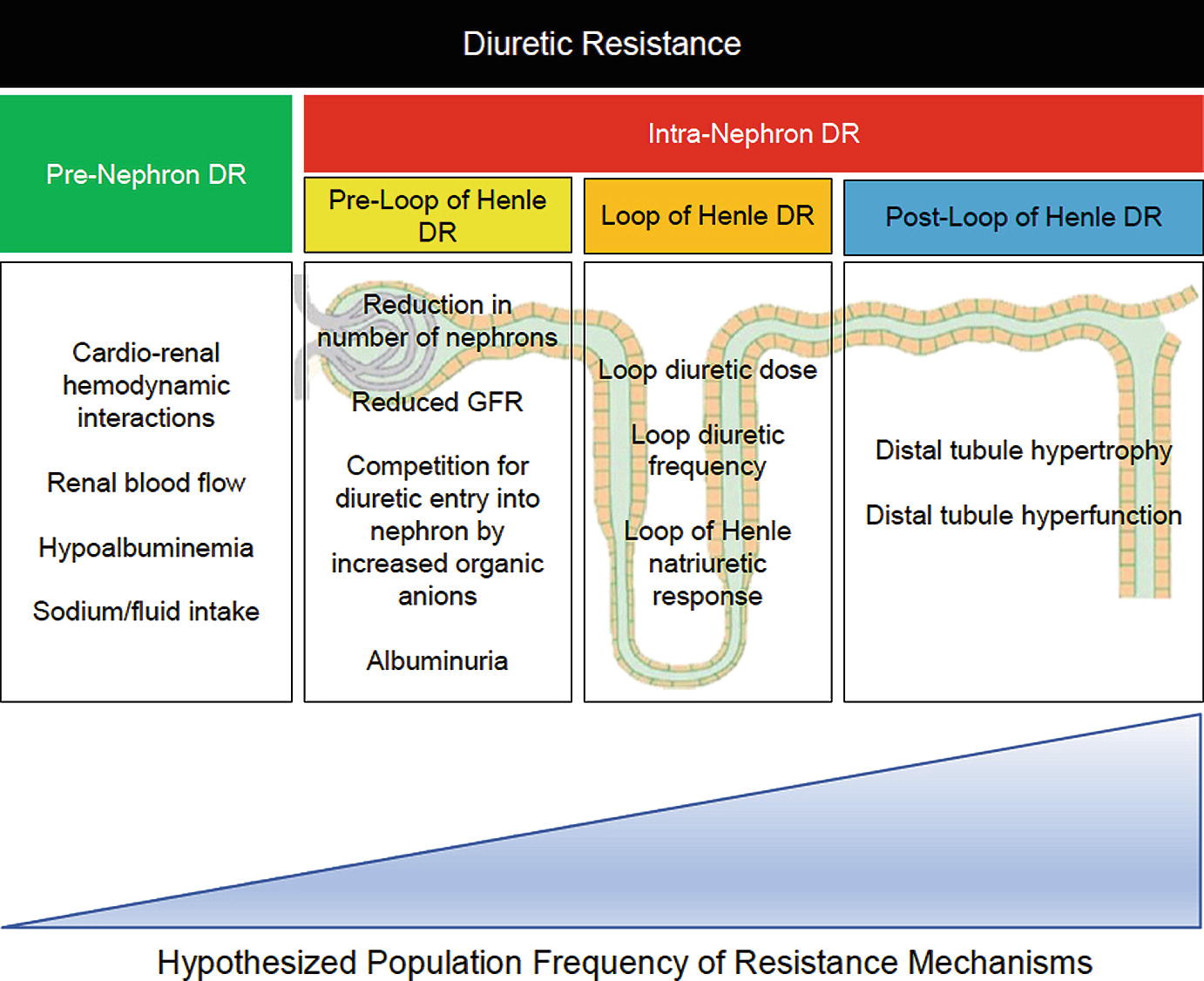
Mechanisms of diuretic resistance
Pre-nephron Diuretic Resistance
Historical literature focusing on pre-nephron and pre-loop of Henle diuretic resistance was performed in healthy subjects or cohorts with hypertension or chronic kidney disease [38–44]. The application of these findings to the AHF patient on modern medical therapies is uncertain. Cardiorenal hemodynamics represent a potential mechanism of pre-nephron diuretic resistance. Poor cardiac output to the kidney was initially believed to be the predominant driver of cardiorenal syndrome and diuretic resistance, but multiple recent analyses have since illustrated that cardiac output is not the primary driver on a population level [13, 14, 45]. Venous congestion, often approximated by elevated right atrial pressure or high intra-abdominal pressure, has been proposed to contribute to diuretic resistance through a reduction in the arterial to venous pressure gradient at the glomerulus. An analysis of the ESCAPE found no difference in baseline right atrial pressure, pulmonary arterial wedge pressure, or cardiac output between patients experiencing high or low diuretic efficiency [20]. Vasodilators such as nesiritide, ularitide, serelaxin, and milrinone failed to augment diuresis by metrics of urine output, IV loop diuretic duration of use, or weight loss in patients with AHF [46–50]. Dopamine at 2 μg/kg/min did not increase urine output compared to placebo in a trial of AHF patients undergoing decongestion with IV loop diuretics [48]. Dopamine trended toward increasing urine volume in those with a baseline systolic blood pressure less than 114 mmHg [48]. Higher diastolic blood pressure predicted greater urine output in the ASCEND-HF trial, although possibly driven by confounding by indication [46]. To the contrary, RAAS antagonism, even in the setting of a blood pressure reduction, may actually improve the natriuretic response to a loop diuretic [51, 52]. Activation of the RAAS varies greatly during decongestion with both diuretics and mechanical volume removal and has unclear associations with diuretic resistance [33, 53]. It remains unclear which patients with lower mean arterial pressures and diuretic resistance should have a temporary cessation in medications that lower blood pressure with initiation of dopamine at 2 μg/kg/min versus those in whom RAAS antagonists should be continued or uptitrated.
Hypoalbuminemia was considered as a pre-nephron diuretic resistance mechanism because all loop diuretics are >90% bound to albumin [54–56]. Hypothesized mechanisms include a reduced intravascular volume available for diuresis and decreased delivery of loop diuretics to the nephron [57]. The majority of literature evaluating the benefit of IV albumin replacement with IV furosemide was performed in nephrotic syndrome or cirrhosis utilizing IV furosemide doses of only 40 mg [41, 58, 59]. A small, retrospective study of patients with a serum albumin <3 g/dL hospitalized for AHF found no difference in net urine output nor diuretic doses needed for diuresis compared to patients with normal serum albumin concentrations [60]. Analysis of the DOSE-AHF and the Renal Optimization Strategies Evaluation in Acute Heart Failure (ROSE-HF) trials found no association between baseline serum albumin concentrations and weight loss (p = 0.43), diuretic efficiency (p = 0.53), or freedom from congestion (p = 0.30) at 72 h [61].
The relationship between sodium and heart failure outcomes is complex [62]. The traditional paradigm teaches that high sodium intake is a driver of pre-nephron diuretic resistance [38, 63]. However, several investigations in AHF populations indicate that a higher sodium intake might be beneficial if a greater net sodium removal is achieved [64]. Hypertonic saline administered with high-dose loop diuretics produced greater urinary sodium excretion, urine volume, and faster achievement of euvolemia than high-dose loop diuretics alone among heart failure patients failing oral combinations of loop and thiazide diuretics [65–67]. However, the quality of data supporting this approach is limited, and it cannot be recommended at this time [68]. Likewise, there is insufficient evidence to recommend any specific dietary sodium limitation for patients with AHF undergoing diuresis [64]. Lastly, non-steroidal anti-inflammatory drugs inhibit renal prostaglandin synthesis leading to a decreased renal blood flow and impaired natriuresis [69, 70].
Pre-loop of Henle Diuretic Resistance
Initially believed to be significant contributors to diuretic resistance from impaired drug delivery to the site of action, renal function and albuminuria are less influential mechanisms of diuretic resistance compared to tubular handling of sodium. In the novel “The House of God”, we see renally-based diuretic adjustments taught with “age + BUN = Lasix dose” [71], which contemporary medical pocket resources continue [72]. Proposed diuretic resistance mechanisms of renal dysfunction are listed in Fig. 11.1. Renal dysfunction is a mechanism of pre-loop of Henle diuretic resistance in chronic kidney disease populations, but is less relevant in the contemporary heart failure patient prescribed an adequate diuretic dose. Estimated glomerular filtration rate (eGFR) poorly correlates with net fluid output (r2 = 0.0; p = 0.35) and diuretic efficiency (r2 = 0.02; p < 0.001) in the ESCAPE [20]. BUN was significantly associated with low diuretic efficiency (OR 1.19 per 10 g/dL increase in BUN; p = 0.005) in a multivariate model incorporating eGFR, which could be a result of increased neurohumoral activation and/or competition with loop diuretic entry into the nephron. Similarly, elevated BUN but not reduced eGFR predicted urine output in the ASCEND-HF trial [46]. When evaluating the relative importance of diuretic delivery and renal tubular response, eGFR did not predict 6 h net fluid or net sodium output [73]. Patients with a low eGFR had decreased diuretic delivery to the kidney, with eGFR (r = 0.58; p = 0.001) and urea clearance (r = 0.75; p = 0.001) showing strong correlation with urinary diuretic concentration. However, patients with lower eGFR compensated for decreased diuretic concentrations by producing greater fractional excretion of sodium at 6 h. Renal function in heart failure has no impact on the individual nephron’s filtrate, but does influence total natriuresis, although at a less significant degree than loop of Henle and post-loop of Henle diuretic resistance. In summary, renal dysfunction is much less of an important mediator of diuretic resistance in AHF than traditional teaching implies.
Albuminuria might contribute to diuretic resistance by binding loop diuretics in the urine as they do in the serum, decreasing the quantity of free drug able to bind to the Na+/K+/2Cl− symporter [72]. Literature supporting this hypothesis was performed in animal models of nephrotic syndrome [74–76], raising questions of its relevance in AHF. Patients with AHF and either normal albuminuria (45%), microalbuminura (42%), or macroalbuminuria (13%) exhibit very weak correlation between diuretic efficiency and urinary albumin concentrations [77]. Recent analyses in humans with nephrotic syndrome also refute albuminuria as a primary mechanism of diuretic resistance, making it implausible that albuminuria is a significant mechanism of diuretic resistance in AHF patients [78].
Loop of Henle Diuretic Resistance
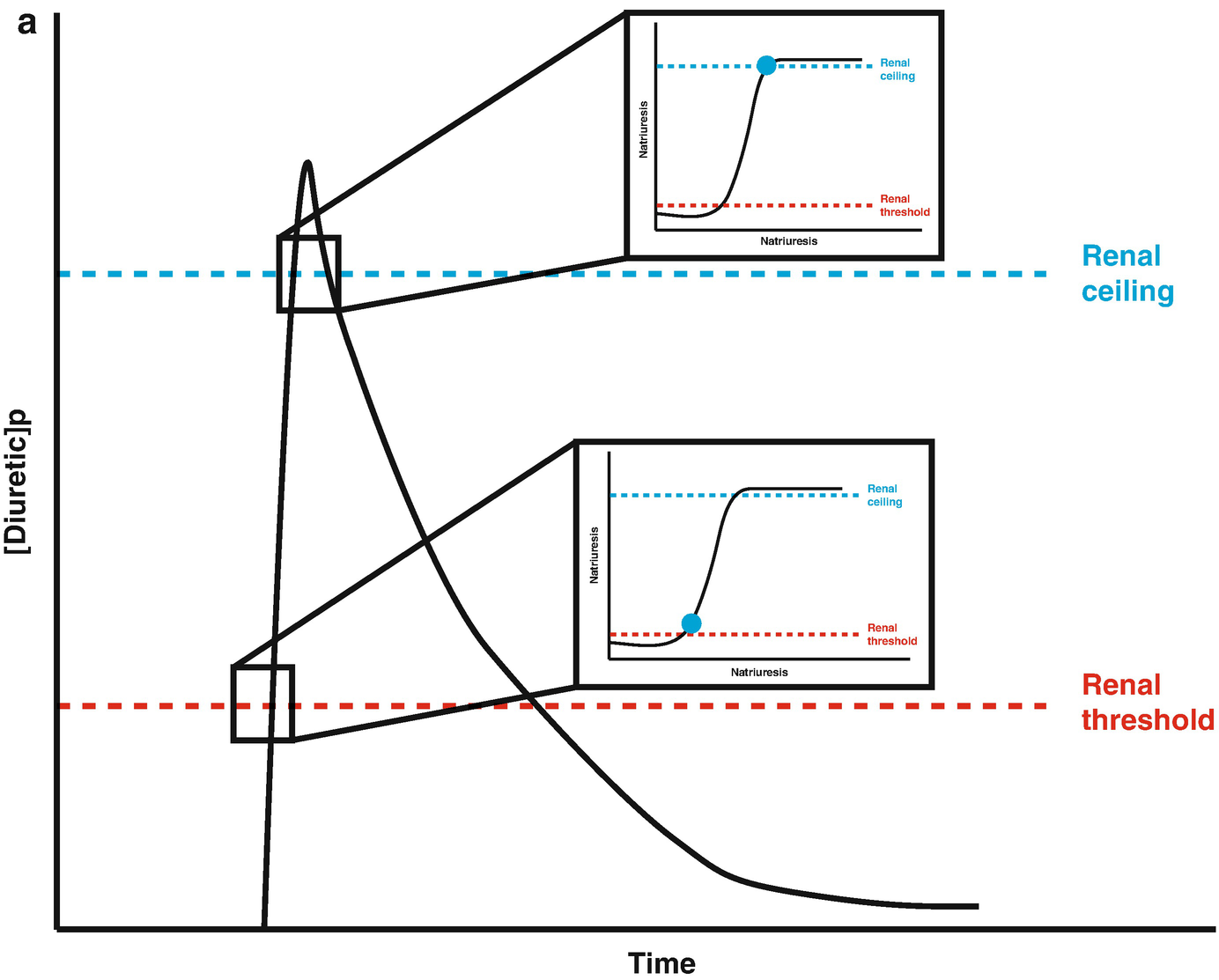
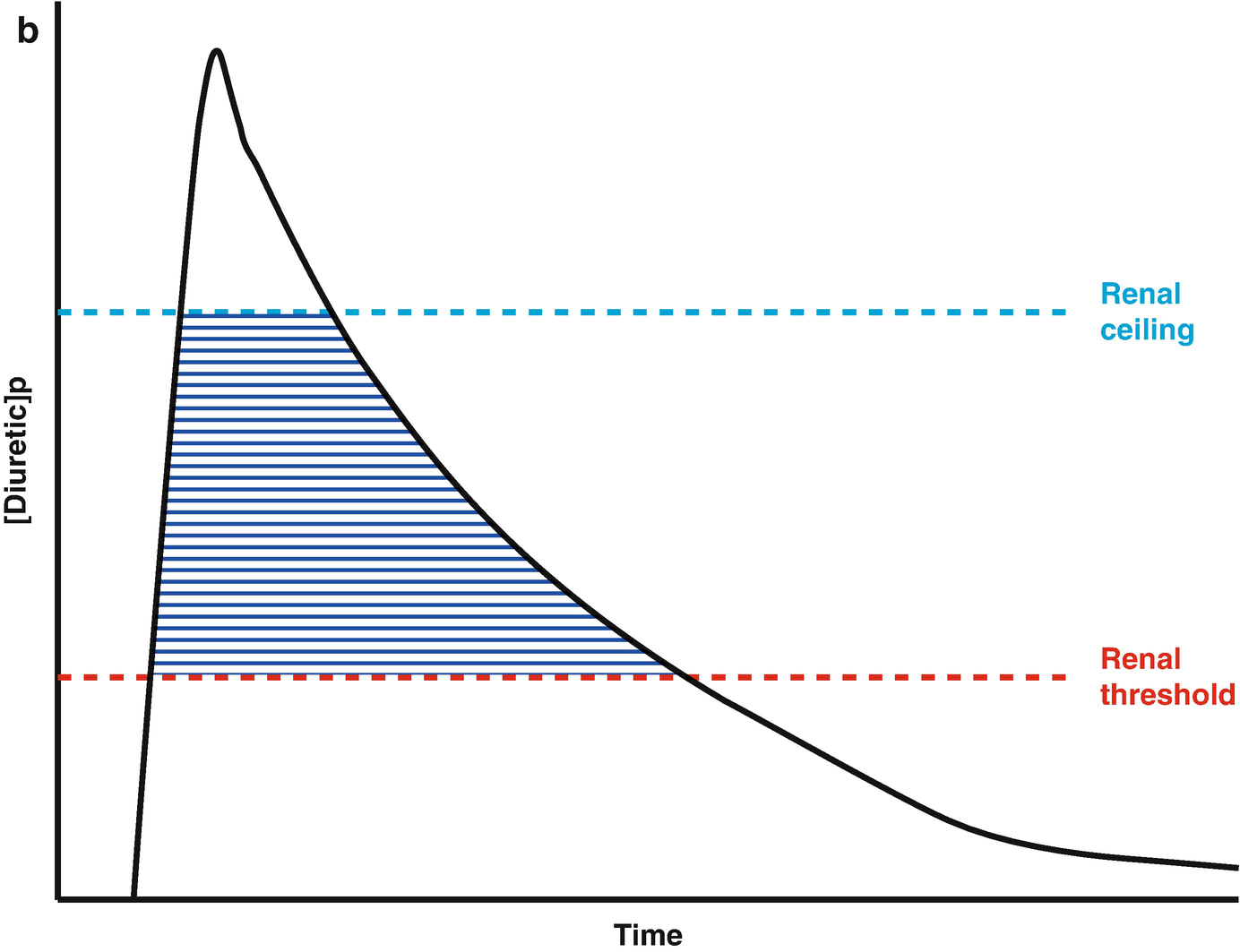
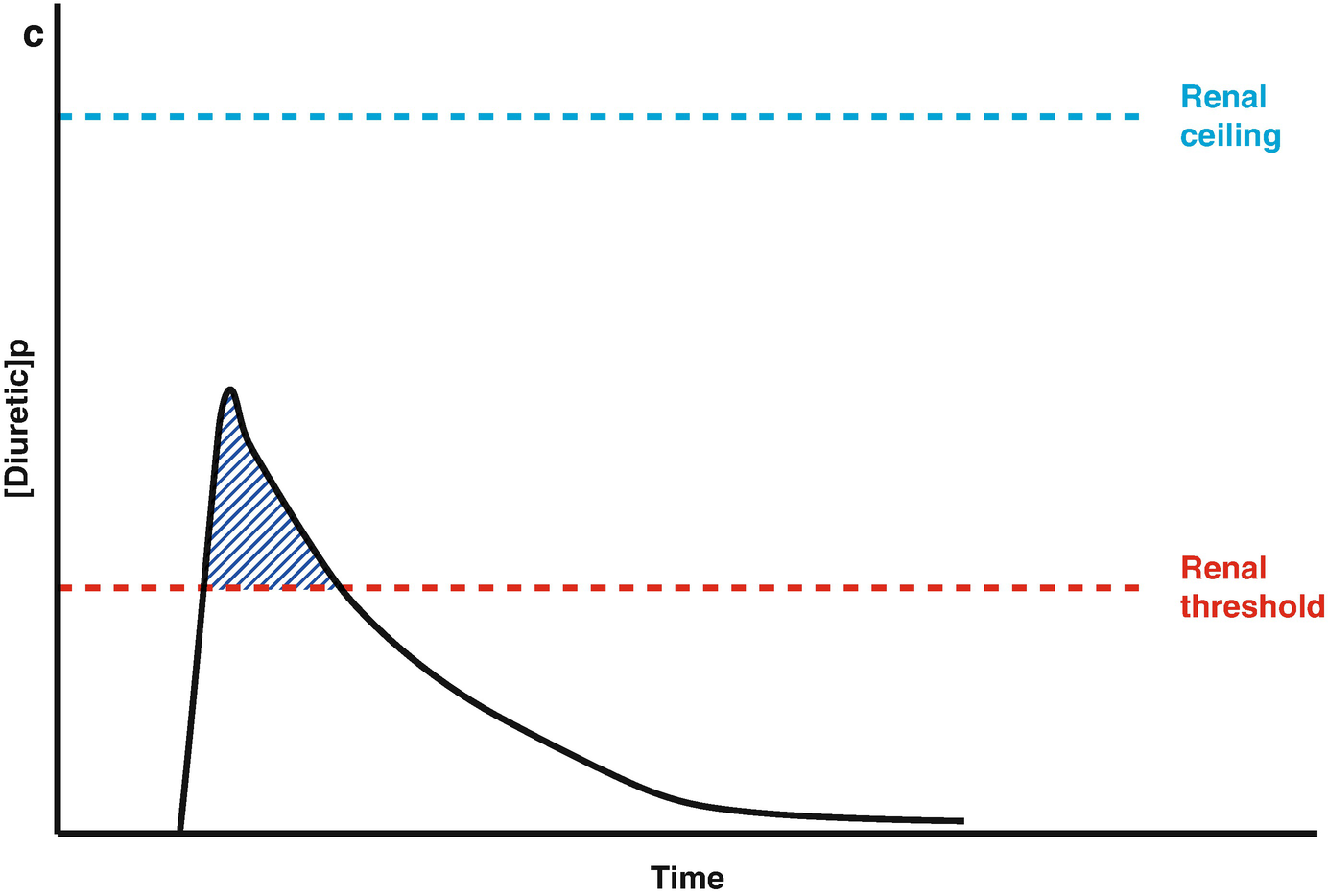
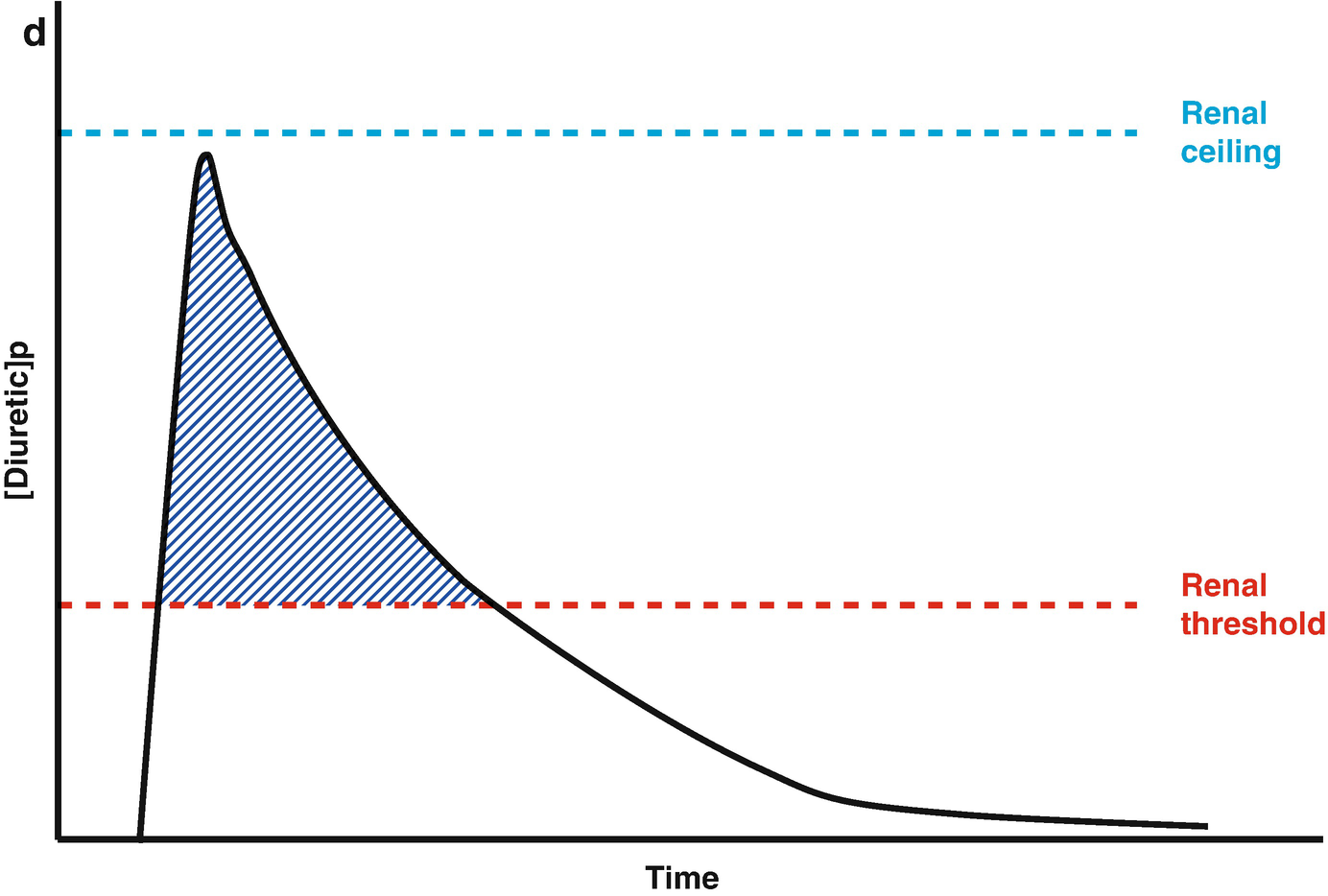
Loop diuretic pharmacokinetics. (a) The loop diuretic plasma concentration (y-axis) is plotted over time (x-axis) when given as an intravenous bolus. The Renal Threshold (red dotted line) is the diuretic concentration that must be exceeded to cause diuresis. The Renal Ceiling (blue dotted line) is the diuretic concentration above which no further increases in diuretic response are gained. The two boxes encasing the moment the diuretic concentration crosses the Renal Ceiling and Renal Threshold illustrates the sigmoidal dose-response relationship between the diuretic concentration (x-axis) and the natriuretic response (y-axis), with the blue dot representing the current loop diuretic concentration on this curve. (b) The loop diuretic plasma concentration (y-axis) is plotted over time (x-axis) when given as an intravenous bolus. The shaded area illustrates the area of the curve between the Renal Threshold and Renal Ceiling. While no further diuretic action is gained when a dose produces a diuretic concentration exceeding the Renal Ceiling, the total volume of diuresis can be increased as a function of maintaining a loop diuretic plasma concentration above the Renal Threshold for a greater time. (c) The loop diuretic plasma concentration (y-axis) is plotted over time (x-axis) when given as an intravenous bolus. The shaded area illustrates the area of the curve above the Renal Threshold. In this scenario, the diuretic response is minimal because of the limited time spent above the Renal Threshold. (d) The loop diuretic plasma concentration (y-axis) is plotted over time (x-axis) when given as an intravenous bolus. The shaded area illustrates the area of the curve above the Renal Threshold. By increasing the loop diuretic dose from Fig. 11.2c, the time above the Renal Threshold was increasing, causing a greater diuretic response
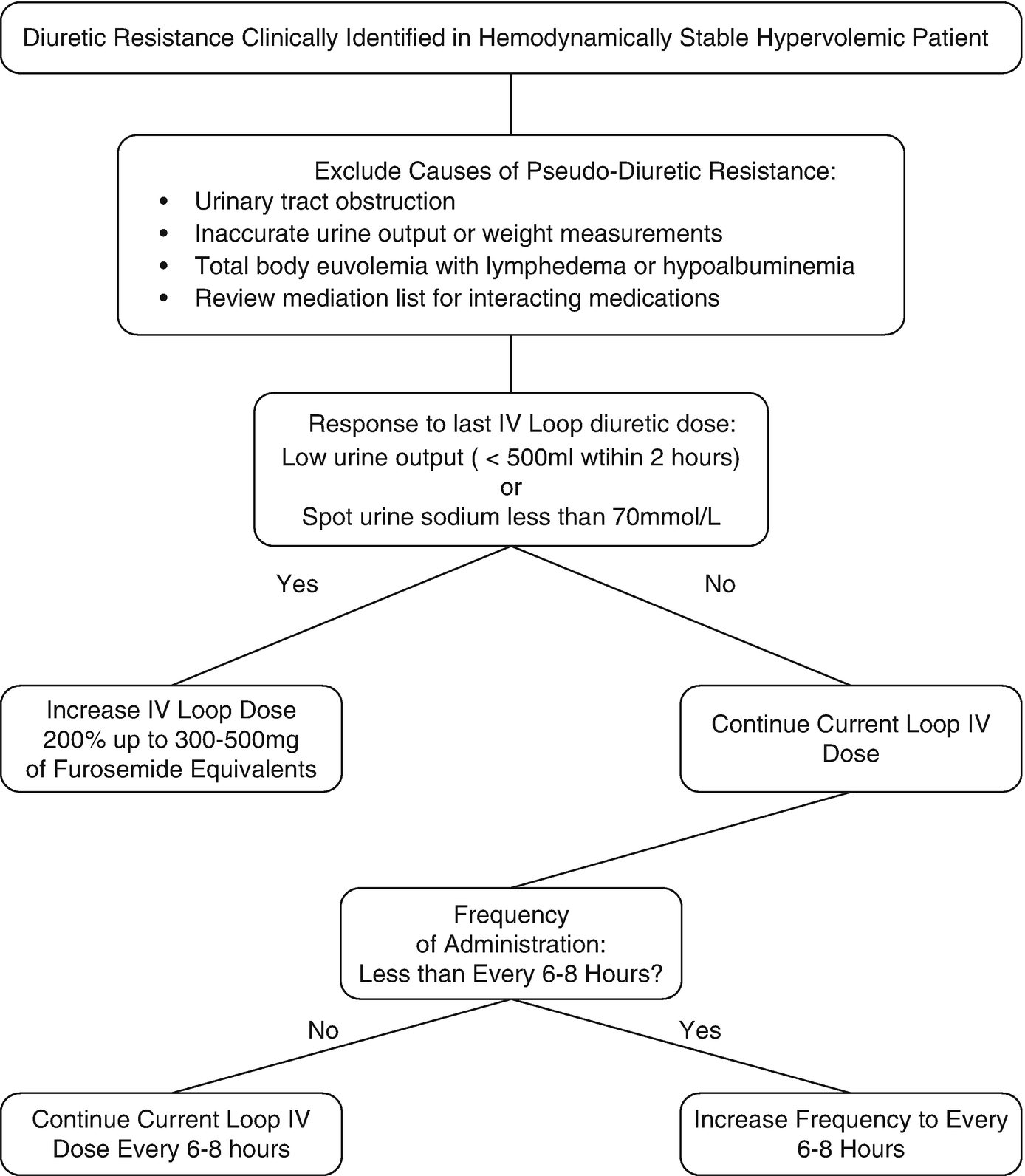
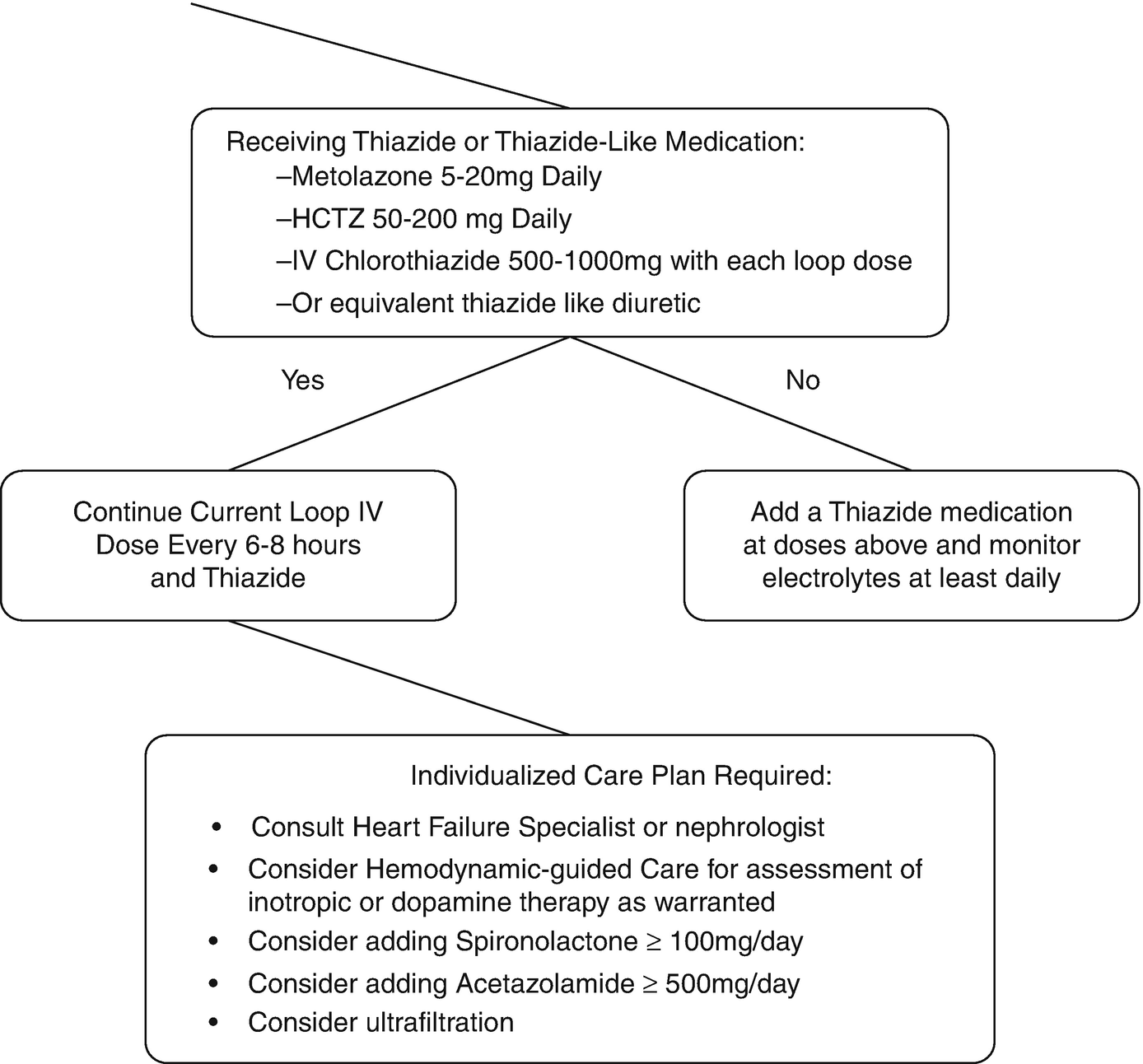
Clinical decision process for addressing loop diuretic resistance
Post-loop of Henle Diuretic Resistance
The few contemporary studies in AHF patients indicate that the majority of diuretic resistance is primarily mediated by post-loop of Henle diuretic resistance. Compared to a pre-diuretic baseline, a median dose of IV furosemide 160 mg (40–270 mg) increased the amount of sodium estimated to be leaving the loop of Henle by 12.6 ± 10.8% (p < 0.001) [83]. Yet, the fractional excretion of sodium only increased 4.8 ± 3.3%, indicating 66% (25–85%) of the sodium leaving the loop of Henle underwent distal tubular reabsorption. After controlling for loop of Henle diuretic resistance by using urine diuretic concentration, the increase in sodium leaving the loop of Henle only accounted for 6.4% of the increase in net fractional excretion of sodium. These findings were substantiated in a study administering IV bumetanide to 50 AHF patients which reported that in the majority (71%) diuretic response was related to intra-renal diuretic resistance via renal tubular changes [73]. Continuous loop diuretic exposure results in rapid distal tubular hypertrophy and hyperfunction in animal models [84–86]. Thus, a focus of restoring diuretic efficacy should be on blocking reabsorption in the distal tubules.
Combination Blockage of the Nephron in Diuretic Resistance
When approaching diuretic resistance, clinicians should first ensure an adequate loop diuretic dose and frequency are prescribed before considering therapeutic strategies targeting post-loop of Henle diuretic resistance (Fig. 11.3). Combination nephron blockade with metolazone has been associated with increased risk of electrolyte abnormalities, worsening renal function, and mortality compared to high-dose loop diuretics, indicating loop diuretics should be maximized as an initial strategy [87].
Thiazide(-like) Diuretics
Thiazide (and thiazide-like) medications are the most commonly utilized medications to overcome loop diuretic resistance [88–90]. Thiazides should be the first option to add to loop diuretics, as they inhibit sodium reabsorption in the distal convoluted tubules where the majority of remaining sodium reabsorption occurs after the loop of Henle. Despite experience spanning 50 years, common misconceptions regarding combining thiazides and loop diuretics persist [89]. Metolazone is often referred to as superior to other thiazide agents, but no solid evidence supports the use of metolazone over other thiazides, even in patients with low eGFR [89, 91–93]. Administration of thiazides 30 min prior to loop diuretics is not evidence-based, as most studies administered both simultaneously [89]. Furthermore, the erratic and delayed absorption profile of metolazone makes this practice irrelevant clinically and unnecessarily increases complexity [94, 95]. IV chlorothiazide has only been compared to oral thiazides in small retrospective studies limiting definitive conclusions on efficacy differences [96]. Any thiazide at equipotent dose [97] in daily or divided doses is appropriate to add to loop diuretic therapy. Careful monitoring for hyponatremia, hypokalemia, and volume status is warranted with combination diuretic therapy to avoid adverse events [89].
Mineralocorticoid Receptor Antagonists
Mineralocorticoid receptor antagonists at diuretic doses are employed with loop diuretics in cirrhotic ascites as the primary combination nephron blockade strategy [98, 99]. In AHF, non-diuretic doses (<50 mg/day) of mineralocorticoid receptor antagonists reduce morbidity and mortality [100], but investigations into larger, diuretic doses have been limited until the Aldosterone Targeted Neurohormonal Combined With Natriuresis Therapy—Heart Failure (ATHENA-HF) trial [101, 102]. The ATHENA-HF trial compared spironolactone 100 mg/day to placebo or continued non-diuretic dose spironolactone in patients with hypervolemic AHF treated with IV loop diuretics [103]. No difference in the primary endpoint of natriuretic peptide change or secondary outcomes such as net urine output, weight change, or titration of IV loop diuretic doses were found [103]. However, ATHENA-HF did not study a population with diuretic resistance and may have utilized insufficient spironolactone doses to achieve therapeutic concentrations of canrenone (active metabolite). Serum potassium levels were no different between spironolactone and placebo, supporting this possibility. Until future studies of diuretic doses of mineralocorticoid receptor antagonists in patients with diuretic resistance are conducted, this class should not be employed over thiazides in combination nephron blockade. Similarly, amiloride cannot be recommended over thiazides in combination nephron blockage because of the reduced capacity for sodium reabsorption in the collecting ducts relative to the distal convoluted tubules.
Vasopressin Antagonists
Vasopressin-2 receptor antagonists have been extensively investigated in AHF, with recent investigations focusing on the diuretic prowess [104]. Three trials comparing tolvaptan to placebo in patients with hypervolemic AHF without diuretic resistance treated with only modest IV loop diuretics found increases in weight loss and urine output [105–107]. However, clinical endpoints such as improvement in dyspnea were not improved, possibly because sodium-free water excretion will have less impact on filling pressures than sodium-rich fluid excretion. Tolvaptan, which acts in the collecting duct, cannot be recommended over thiazides in combination nephron blockage at this time given the limited study in loop diuretic resistance.
Diuretics in the Proximal Tubules of the Nephron
Medications acting in the proximal convoluted tubules such as acetazolamide have shown promise in preliminary investigation of combination nephron blockade and are undergoing further investigation in the Acetazolamide in Decompensated heart failure with Volume Overload (ADVOR) trial [108, 109]. Acetazolamide may be a promising diuretic to add when diuretic resistance persists despite combination nephron blockade with loop diuretics and thiazides but there is no conclusive evidence up to date.
In conclusion, diuretic resistance is problematic to universally define but prevalent in AHF. The mechanisms driving diuretic resistance are diverse and the subsequent strategies should be specific to the mechanism. Optimization of loop diuretic regimens should be the primary strategy followed by combination nephron blockage with thiazides. Several additional strategies are promising but require further investigation before they can be recommended.
Treatment Pearls for the Case Vignette
In the case vignette, a first step would be to increase the IV furosemide dose to 200–240 mg IV bolus to overcome potential loop of Henle diuretic resistance given the low urinary output of the patient with 120 mg. In addition, it is reasonable to increase frequency of furosemide bolus dosing up to every 6 or 8 h in an attempt to overcome post-diuretic sodium retention.
Combination nephron blockage should be considered if the diuretic response is not restored on furosemide 240 mg IV every 6 to 8 h. Medical options then include thiazide diuretics, which block sodium reabsorption in the distal convoluted tubules and have the most empirical evidence in support. Alternatives such as mineralocorticoid receptor antagonists, epithelial sodium channel antagonists such as amiloride, vasopressin antagonists and proximal-working diuretics might be employed in individual cases.

Full access? Get Clinical Tree


