Primary Versus Secondary Dilated Cardiomyopathy
Dilated cardiomyopathy (DCM) is heart disease usually characterized by four-chamber dilatation in the absence of significant valvular, ischemic, or hypertensive disease. In the past, some have referred to ischemic heart disease as a form of DCM; hence, the generic division between “ischemic” and “nonischemic” cardiomyopathy is frequently made clinically.
1 However, it is important to note that while ischemic heart disease may cause ventricular dilatation, it should not be considered a cardiomyopathy. Moreover, despite the frequent use of the term “nonischemic dilated cardiomyopathy,” it is considered both nonspecific and redundant and is therefore not accepted among pathologists.
2,3In the past, DCM was divided into “idiopathic,” familial, and secondary types. Currently, it is understood that genetics plays a role in the majority of cases of DCM, even though a family history is elicited in only one-third.
4 There is no clear distinction between a cause and an association, blurring the lines between primary and secondary cardiomyopathy. For example, alcoholism may be a risk factor but not necessarily a cause for cardiomyopathy, and myocarditis may play a role in the development of heart disease in patients with cardiomyopathic mutations. For these reasons, there is a trend away from the designation “idiopathic dilated cardiomyopathy” as the line between genetic and environmental blurs.
In general, the term DCM should be employed by pathologists when no secondary factor is identified to explain the pathology.
5 However, if a nongenetic condition (e.g., myocarditis, hemochromatosis, etc.) is identified, the condition is best characterized as heart disease secondary to such (e.g., myocarditis-associated heart disease, cardiac hemochromatosis, etc.).
Tables 156.1 and
156.2 present two currently accepted classifications of cardiomyopathy, and
Table 156.3 a functional pathologic classification.
Phenotype-Genotype Correlation
Sampling limitations of endomyocardial biopsy, biases of autopsy studies, and lack of specificity of histologic findings have hampered the development of a pathologic classification of cardiomyopathy, especially DCM. It was hoped in the early years of this century that molecular studies would reveal a good correlation between classes of mutated proteins and type of cardiomyopathy as defined by imaging, hemodynamics, and electrocardiographic findings. However, molecular investigations of patients with cardiomyopathy have resulted in a large number of mutations that lack specificity for a specific clinical type and have thus far shown little correlation between phenotype and genotype. The American Heart Association classification, which relied heavily on genetics, was criticized by the European Society of Cardiology, who continued to rely primarily on morphology
6,8 (
Table 156.2).
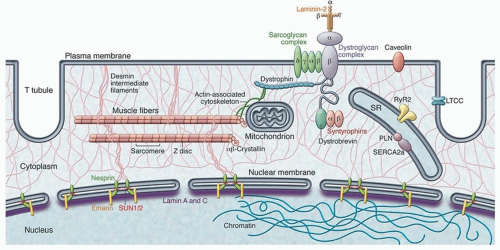
The major genes implicated in the pathogenesis of DCM include those encoding for proteins in the sarcomere (e.g., myosin heavy chain and titin), cytoskeleton (e.g., sarcoglycans and dystrophin), and nuclear membrane (e.g., lamin A) (
Table 156.4;
Fig. 156.1). Mutations in sarcomeric genes were originally discovered in familial hypertrophic cardiomyopathy (HCM) but are also found in DCM.
Currently, there is a movement to use a more descriptive classification system for clinicians, which relies on five criteria. The “MOGES” system uses morphofunctional phenotype (M), organ involvement (O), genetic or familial inheritance pattern (G), etiologic description (E) of genetic defect or nongenetic underlying cause, and the functional status (S) to characterize patients with cardiomyopathy of all types, including DCM.
11From the standpoint of the pathologist, it is important at a gross level to identify the phenotype of cardiomyopathy. There are currently five (
Table 156.3): DCM, HCM, arrhythmogenic cardiomyopathy (AC), restrictive cardiomyopathy (RCM), and left ventricular noncompaction (LVNC). As will be addressed in Chapter 25, of these designations, there is the greatest pathologic variation in RCM, which is a phenotypic manifestation of diverse unrelated conditions.
The current chapter is devoted to DCM in adults, focusing on both the genetic varieties as well as those cases associated with drugs, pregnancy, viruses, and nutritional deficiency. Cardiomyopathy of childhood and those with extracardiac manifestations are discussed in Chapter 23. DCM occurring as a consequence of systemic diseases is discussed in detail in Section 3.
Autopsy Evaluation of Dilated Cardiomyopathy
The autopsy evaluation of cardiomyopathy should begin with review of clinical data. Some cases may have limited or no information (particularly in relatively young individuals who experience sudden death) and others may have extensive clinical and even molecular genetic data
available. In the former scenario, the main purpose of the autopsy is to establish cause of death, (see
Chapter 141), including cardiac arrhythmia, thromboembolism, and extracardiac or nonnatural causes, and to try and identify potential risk factors (hypertension, chronic renal disease, obesity, etc.). In the cases of a medical autopsy with abundant clinical data, the purpose of autopsy generally revolves around aspects of therapy, for example, ventricular assist devices or defibrillators, and the evaluation of extracardiac causes of death, such as infections and thromboembolism.
The first gross heart parameters to establish are the presence of cardiomegaly (preferably using tables based on body weight and height)
12 and the absence of other causes of myocardial disease, such as ischemic, valvular, and hypertensive diseases. In general, a heart weight >50% above the expected mean is in keeping with cardiomegaly. It should be noted that there may be coexistence of true cardiomyopathy with moderate coronary or moderate valve disease, in which case the extent of disease should be estimated.
In the end, it is a judgment call as to whether or not the extent of the underlying diseases is sufficient to explain the observed cardiac phenotype. Evaluation of clinical records, if available, can be helpful in determining the severity of comorbid states such as hypertension and valvular disease. However, evaluating for extracardiac manifestations of the disease can also be helpful. For example, if no hypertensive changes are observed in the kidney, it is unlikely that the hypertension alone could explain cardiomegaly.
Occasionally, the pathologist will be asked to perform molecular testing in cases that have been undertaken during life. Such molecular genetic testing affords the ability to formally characterize a process that cannot otherwise be explained. More importantly, it allows for more complete characterization of a disease that often has heritable implications. Confirming a genotype (or even absence of an identifiable mutation) can be immensely valuable when screening the kindred and assessing risk of surviving family members.
Explant
The diagnosis of cardiomyopathy at cardiac explant is generally straightforward, as the patient has been extensively evaluated prior to transplant and the diagnosis is not in doubt. In some cases, an unexpected diagnosis may be found, if a biopsy has not been performed. For example, a pattern of subepicardial replacement-type fibrosis may be indicative of prior myocarditis. Small foci of active myocarditis (“smoldering myocarditis”) can also be identified. Sarcoidosis and hemochromatosis may also manifest as ventricular dilatation and failure. Although unusual, ischemic heart disease may occasionally be missed clinically, so it is important to carefully evaluate for such.
13 Similarly, there may be areas of extensive fibrofatty replacement of the ventricle, suggestive of a diagnosis of AC (Chapter 21).
Endomyocardial Biopsy
Because of limited sampling and lack of specific histologic features, endomyocardial biopsy is useful in excluding a specific pathologic entity, most commonly sarcoidosis or myocarditis. Ultrastructural evaluation is of limited use in most cases, although a search for membranebound autophagic vacuoles (Danon cardiomyopathy) or lamellar inclusions (Fabry disease) should be performed if indicated, as these conditions may occasionally present as a dilated phenotype (the former more so than the latter, which usually manifests as a hypertrophic phenotype).