18 Dilated Cardiomyopathy
Etiology and Pathogenesis
DCM is characterized by dilatation and impaired contraction of either the left ventricle or both ventricles, as a result of altered structure or function in diseased cardiomyocytes. Before the heart becomes dilated and weak, there is either an index event (e.g., a myocardial infarction [MI] or acute myocarditis) that leads to impaired ventricular contractility, or progression of underlying disease (e.g., severe valvular regurgitation) that leads to ventricular pressure overload causing systolic dysfunction. Because of ventricular systolic dysfunction, gradual compensatory responses of the cardiomyocytes lead to cardiac remodeling (Fig. 18-1). Initially the cardiomyocytes respond by becoming hypertrophied, but the poorly functioning ventricle gradually dilates to handle the progressive volume overload. In most cases, contractility is impaired initially and primarily in the left ventricle, but as systolic dysfunction progresses, the right ventricle also becomes enlarged and hypokinetic. Rarely, the cardiomyopathic process will affect primarily the right ventricle at the outset of the DCM.
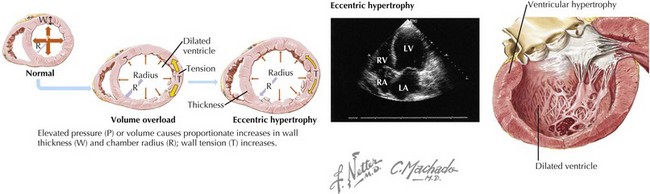
Figure 18-1 Cardiac remodeling secondary to volume overload.
LA, left atrium; LV, left ventricle; RA, right atrium; RV, right ventricle.
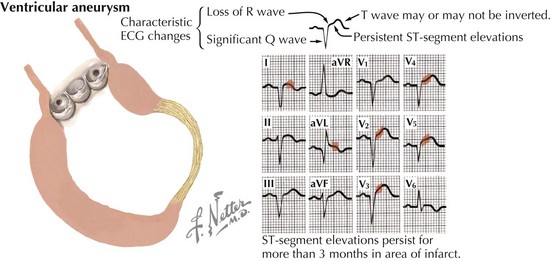
Of the many causes of DCM (Table 18-1), the most common in the United States is ischemic heart disease. After an MI, the infarct scar may expand to develop into a large area of nonfunctioning myocardium during the first hours and days after an acute MI. During this time, left ventricular (LV) systolic function may be maintained by hypercontractility of the noninfarcted portion of the left ventricle. Longer term, over days to months to years, global remodeling occurs, resulting in a dilated and poorly contractile ventricle. In some cases, a ventricular aneurysm may form (Fig. 18-2). Because coronary artery disease (CAD) is such a frequent cause of DCM—contributing to approximately two thirds of all cases of HF—the nomenclature for DCM is often subdivided into ischemic cardiomyopathy (ICM) versus nonischemic cardiomyopathy. To be classified as an ICM, the burden of coronary disease must be in proportion to the systolic dysfunction. The definition of ICM is thus based on systolic dysfunction in patients with a history of MI, patients who have undergone revascularization procedures (coronary artery bypass surgery or percutaneous coronary intervention), patients with 75% or greater stenosis of the left main or proximal left anterior descending artery, and patients with 75% or greater stenosis of two or more epicardial vessels.
Table 18-1 Etiologies of and Evaluation for Dilated Cardiomyopathy
| Etiology | Targeted Evaluation |
|---|---|
| Ischemic heart disease (coronary artery disease) | Coronary angiography (gold standard), noninvasive coronary imaging (CT or MRI), stress test |
| Hypertension* | Physical examination (not helpful when end-stage) |
| Valvular heart disease | Physical examination, echocardiography, cardiac MRI |
| Infectious (e.g., viral; Chagas disease; Lyme disease) | |
| Cardiotoxins (e.g., alcohol, anthracycline; excess catecholamines; heavy metals—lead, arsenic, cobalt) | |
| Metabolic/endocrine (e.g., hypothyroidism, hyperthyroidism; diabetes mellitus; acromegaly; adrenal insufficiency; pheochromocytoma) | |
| Connective tissue disease (e.g., systemic lupus erythematosus; scleroderma*; dermatomyositis; polyarteritis nodosa; rheumatoid arthritis) | ANA ± ENA and other specific rheumatologic markers |
| Infiltrative (e.g., Wilson’s disease; sarcoidosis*; hemochromatosis*; amyloidosis*) | |
| Metabolic/nutritional (e.g., magnesium deficiency; kwashiorkor; anemia; beriberi; selenium deficiency) | |
| Peripartum cardiomyopathy | Temporal relationship to pregnancy |
| Giant cell myocarditis | Endomyocardial biopsy |
| Muscular dystrophies (e.g., Duchenne; Becker-type; myotonic dystrophies) | Genetics |
| Familial (e.g., X-linked) | Family history, genetics |
| Idiopathic | (Diagnosis of exclusion) |
ACE, angiotensin-converting enzyme; ANA, anti-nuclear antibody; CBC, complete blood count; ENA, extractable nuclear antigens; GH, growth hormone; HbA1c, hemoglobin A1c; Ig, immunogobulin; PCR, polymerase chain reaction; SPEP, serum protein electrophoresis; TSH, thyroid-stimulating hormone; UPEP, urine protein electrophoresis.
* Diseases that can belong to more than one type of cardiomyopathy (e.g., hypertrophic or restrictive).
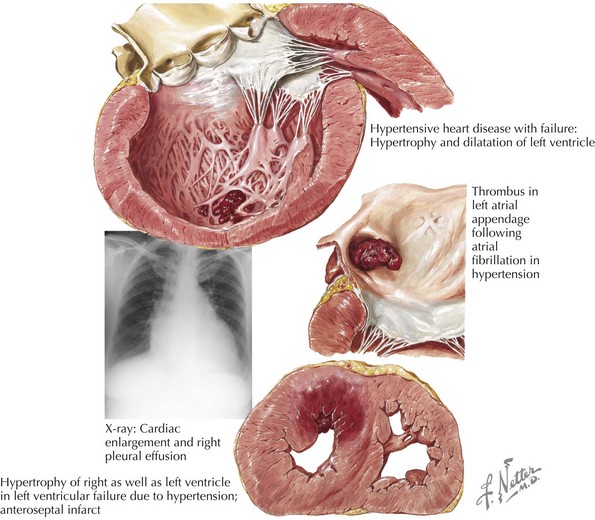
Other common etiologies for DCM are end-stage hypertensive heart disease (Fig. 18-3) and valvular heart disease (“valvular cardiomyopathy”). Less common etiologies include cardiotoxins such as alcohol and anthracycline and herceptin chemotherapies; abnormal metabolic state or endocrinopathies such as thyroid disease, diabetes, acromegaly, adrenal cortical insufficiency, pheochromocytoma; autoimmune diseases such as connective tissue diseases (e.g., scleroderma, systemic lupus erythematosus) and giant cell myocarditis; infiltrative diseases such as sarcoidosis, hemochromatosis, and amyloidosis; nutritional deficiencies; peripartum state; and familial/genetic diseases (e.g., muscular dystrophies, MELAS [mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke] syndrome, and other recently discovered associated chromosomal abnormalities).
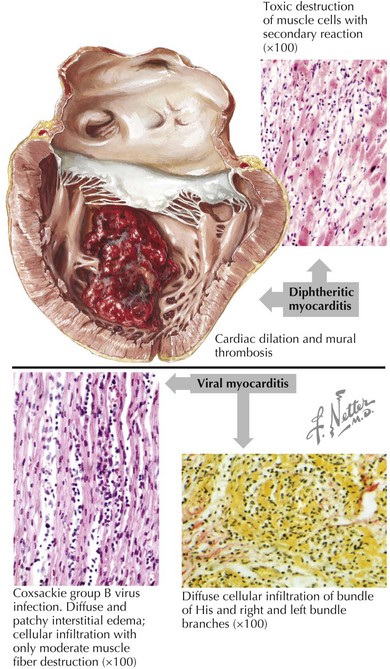
When the etiology is thought to be an infectious agent because of a viral prodrome, the specific pathogen is often not identified, in which case the generic term “viral myocarditis” is commonly used. Histologically there is usually a diffuse inflammatory response with lymphocytes infiltrating the myocardium (Fig. 18-4). Specific pathogens that have been associated with DCM development include viruses such as Coxsackie B virus, enterovirus, adenovirus, parvovirus, HIV, and cytomegalovirus; and parasites such as trypanosomiasis in Chagas disease (the most common cause of infectious cardiomyopathy in South America) and Lyme disease. Although no specific bacterium or fungus has been known to cause cardiomyopathy, acute ventricular systolic dysfunction has been seen in the setting of sepsis, presumably due to the effect of endotoxins or other mediators.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


