Digitalis Glycosides
Paul J. Hauptman
Ralph A. Kelly
Historical Perspectives
Cardiac glycosides have played a prominent role in the therapy of congestive heart failure since William Withering codified their use in his classic monograph on the efficacy of the leaves of the common foxglove plant (Digitalis purpurea) in 1785. Nevertheless, a controversy has existed about whether the risks of digitalis preparations outweigh their benefits, particularly in patients with heart failure in sinus rhythm (1). The standard for clinical use of the cardiac glycosides in modern medicine was reflected in a debate between two eminent clinicians, who were also the co-editors of The Oxford Medicine, Henry Christian and James Mackenzie. Mackenzie advocated the use of digitalis preparations only in those patients with heart failure who also had atrial arrhythmias, prompting the following response from Christian (2):
My views evidently differ from those of my fellow editor of the Oxford Medicine. The views of Sir James MacKenzie have been concurred in by numerous observers, with the result that there is a growing feeling that, unless the pulse is absolutely irregular and rapid, little is to be gained from digitalis therapy. My own experience is so directly contrary to this that is seems worthwhile to restate the views already expressed by me.…My own view with regard to digitalis, as a rule, is that it has a striking effect on those changes in the patient which are brought on by cardiac insufficiency, and this effect appears irrespective of whether or not the pulse is irregular.
Within the United States, at least, Christian’s views prevailed until several retrospective and uncontrolled trials in the 1970s pointed to a lack of efficacy of digitalis preparations in many patients for whom they were prescribed. This debate became more relevant with the availability of other less-toxic remedies for heart failure, most notably the introduction of loop diuretics in the 1960s, the development of new classes of vasodilators in the 1980s, and the advent of the β-adrenergic antagonists (β-blockers) in the 1990s. Although a number of prospective controlled trials (discussed in detail later) that document the safety and efficacy of digitalis in patients with moderate and severe congestive heart failure have been published, controversy over the role of digoxin gained intensity with the recognition that serum digoxin levels may be important and the persistence of uncertainty about the potential lack of clinical efficacy in certain patient subgroups.
In this chapter, we begin with a brief overview of the basic pharmacology of the actions of cardiac glycosides. This is followed by a review of the pharmacokinetics of digoxin with important drug interactions. The data justifying the use of digoxin in patients with moderate to severe congestive heart failure are reviewed in some detail, followed by a description of the mechanisms underlying the toxic manifestations of this class of drugs and their treatment. We also offer a section on contemporary practice that includes a discussion of the place digoxin has in the published guidelines of various professional societies. Readers interested in citations prior to 1990, particularly those citations that address the basic pharmacology of the cardiac glycosides, are referred to the first edition of this text.
Cardiac Glycosides: Natural Sources and Chemical Structure
The terms digitalis and cardiac glycosides are used in this chapter to refer to any of the steroid or steroid glycoside
compounds that exert characteristic positive inotropic and electrophysiological effects on the heart. Although there are important differences in pharmacokinetics among the more than 300 known compounds with these properties, their pharmacologic actions are fundamentally similar. They are found in plants and in the venom and skin of certain toads. Clinically useful preparations are derived from the leaves and seeds of plants in the genera Digitalis and Strophanthus. Digitoxin is derived from the leaves of D. purpurea, and digoxin is derived (after a mild alkaline hydrolysis step) from those of D. lanata, as are lanatoside C and deslanoside. From the seeds of S. gratus comes ouabain, a hydrophilic, relatively rapidly acting cardiac glycoside. Currently, digoxin is by far the most commonly prescribed of these drugs due to the ready availability of techniques for measuring serum levels, flexibility in its routes of administration, and its intermediate duration of action.
compounds that exert characteristic positive inotropic and electrophysiological effects on the heart. Although there are important differences in pharmacokinetics among the more than 300 known compounds with these properties, their pharmacologic actions are fundamentally similar. They are found in plants and in the venom and skin of certain toads. Clinically useful preparations are derived from the leaves and seeds of plants in the genera Digitalis and Strophanthus. Digitoxin is derived from the leaves of D. purpurea, and digoxin is derived (after a mild alkaline hydrolysis step) from those of D. lanata, as are lanatoside C and deslanoside. From the seeds of S. gratus comes ouabain, a hydrophilic, relatively rapidly acting cardiac glycoside. Currently, digoxin is by far the most commonly prescribed of these drugs due to the ready availability of techniques for measuring serum levels, flexibility in its routes of administration, and its intermediate duration of action.
A detailed description of sources of cardiac glycosides, their chemistry, and structure-activity relationships are extensively considered in standard texts (3,4). The steroid nucleus common to all cardiac glycosides contains an α, β-unsaturated lactone ring attached at the C-17 position. Without the sugar moieties, the steroid and unsaturated lactone part of the molecule is called a genin or an aglycon. The genins are usually less potent and have more transient actions due to altered pharmacokinetics than do the parent glycosides. It is apparent from structure-activity comparisons that the cardioactive glycosides demonstrate considerable variation in the structure of the steroid nucleus, as well as the sugar substituents attached at the C-3 position. Digitoxin differs from digoxin only by the absence of a hydroxyl group at C-12. This absent hydroxyl group reduces the hydrophilicity of the compound and results in markedly different metabolism and protein binding compared with digoxin.
Mechanism of Action
Positive Inotropic Effect
By the late 1920s, it became clear that digitalis preparations caused a positive inotropic effect on the intact ventricle, resulting in an increase in the rate of increase of intracavitary pressure during isovolumic systole at constant heart rate and aortic pressure. This effect could be demonstrated in normal, as well as failing, cardiac muscle. Cardiac glycoside administration caused the ventricular function (Frank-Starling) curve of the intact heart to shift upward and to the left, so that more stroke work is generated at a given filling pressure. This was found to be true of both right and left ventricles, and of atrial as well as ventricular myocardium. Force-velocity curves for isolated cardiac muscle are shifted in parallel upward and to the right by cardioactive steroids. These effects appear to be sustained during in vivo administration of digitalis, for periods of weeks to months, without any evidence of desensitization or tachyphylaxis. The time to peak force generation and the relaxation rates are altered little by subtoxic doses or concentrations of cardioactive steroids, but the positive inotropic effect observed is highly dependent on contraction frequency, declining on either side of an intermediate frequency that yields the maximal inotropic response.
It is now generally believed that digitalis compounds bring about an increase in the availability of activator Ca2+ in heart cells and that this increase in intracellular Ca2+ activity is sufficient to explain both the inotropic and arrhythmogenic effects of these drugs (3,5,6). Importantly, it is also now accepted that the increase in intracellular Ca2+ is a consequence of the direct effect of cardiac glycosides on transmembrane Na+ transport (3,4,5,6,7).
Inhibition of Na,K-ATPase
All cardioactive steroids share the properties of being potent and highly specific inhibitors of the intrinsic membrane protein Na,K-adenosine triphosphatase (Na,K-ATPase). The plasma membrane Na,K-ATPase, the molecular machinery that comprises the cellular sodium pump, is representative of a family of evolutionarily ancient enzymes in which membrane ion translocation is coupled to the hydrolysis of a high-energy ATP phosphate. Indeed, Jens Skou of Denmark was awarded the 1997 Nobel Prize in Chemistry for his discovery in the 1950s and 1960s of the enzyme activity he termed the Na+,K+-ATPase, which he characterized in giant neurons of shellfish (8,9). The α or catalytic subunit component of the sodium pump, the roughly 100-kDa integral membrane protein that almost certainly contains the Na+, K+, and ATP binding sites of the intact enzyme, has been highly conserved in eukaryotes for the establishment and maintenance of transmembrane Na+ and K+ gradients. Unique to the Na,K-ATPase is the presence of an approximately 40-kDa glycosylated subunit termed the γ subunit, the function of which is as yet unknown. This subunit, too, has been highly conserved as indicated by nucleotide sequence analysis from phylogenetically diverse species ranging from the electric fish Torpedo californica to humans (10,11). A third, small hydrophobic subunit termed τ is not necessary for association, membrane targeting, or function of α/β subunits but appears to regulate activation of Na,K pumps by K+ (12,13,14).
Aside from its α,β-heterodimer structure, another characteristic of the Na,K-ATPase that is unique among the P-type ATPases is the presence of a binding site on the extracytoplasmic face of the α subunit for the cardiac glycosides (15,16). Although the binding affinity varies among isoforms of the enzyme and from species to species, the ability of the enzyme to bind and to be inhibited by cardiac glycosides has been very highly conserved; it is so highly conserved, in fact that cardiac glycoside binding is a sine qua non for identifying functional Na,K-ATPase and is used to define the contribution of the sodium pump to plasmalemmal monovalent cation fluxes. One cardiac glycoside binding site facing the extracellular surface is present per α chain. Optimal binding requires Na+, Mg2+, and ATP and is inhibited by extracellular K+. Cardiac glycoside binding results in complete inhibition of enzymatic and transport functions of each Na,K-ATPase site occupied.
In addition to a wealth of evidence from animal, whole heart, isolated muscle, single cell, and molecular studies, inhibition of the sodium pump in atrial tissue of patients
treated with conventional doses of digoxin also has been demonstrated. Indeed, the highly selective action of the cardiac glycosides to bind to the digitalis binding site on the extracellular face of the α subunit of Na,K-ATPase has engendered much speculation about the possible existence of endogenous ligands, simply because the amino acid sequence and conformation that form their binding site has been so highly conserved over many phyla and millennia. Although many intriguing data exist, there is, as yet, no proof that any endogenous digitalis exists that has a well-defined biological role in regulating Na,K-ATPase function.
treated with conventional doses of digoxin also has been demonstrated. Indeed, the highly selective action of the cardiac glycosides to bind to the digitalis binding site on the extracellular face of the α subunit of Na,K-ATPase has engendered much speculation about the possible existence of endogenous ligands, simply because the amino acid sequence and conformation that form their binding site has been so highly conserved over many phyla and millennia. Although many intriguing data exist, there is, as yet, no proof that any endogenous digitalis exists that has a well-defined biological role in regulating Na,K-ATPase function.
Three isoforms of the α subunit of Na,K-ATPase have been identified. Several investigators (17,18) have suggested, based on high-resolution immunocytochemical staining techniques using antibodies specific for each isoform, that each α subunit isoform subserves a distinct subcellular role. The α1 subunit appears to regulate bulk cytosolic Na+, whereas α2– and α3-containing Na,K-ATPases appear to be localized to areas of the sarcolemma in close apposition to the endoplasmic reticulum (i.e., sarcoplasmic reticulum in muscle) (18). Because the isoforms differ in their affinities for cardiac glycosides (indeed, it was this property that led to their original discovery), the subcellular targeting of each α subunit isoform may impact importantly on the pharmacology of these drugs.
Na+-Ca2+ Exchange and Increased Intracellular Calcium
Compelling direct evidence supporting cardiac glycoside-induced increases in intracellular Na+ concentration or activity ([Na+]i) is available from studies of cardiac cells impaled with Na+-sensitive microelectrodes. The relationship of [Na+]i to tension development is direct and remarkably steep. Data from these and other experiments support the view that inhibition of active cellular Na+ transport results in augmentation of myocyte Ca2+ content, which, in turn, produces a positive inotropic response. This is analogous to the increase in contractility that follows an increase in [Na+]i due to increased contraction frequency, a phenomenon termed treppe or Bowditch staircase. The mechanism of both digitalis-induced positive inotropy and the staircase phenomenon appears to involve an altered balance between intracellular Na+ and Ca2+. The transmembrane Na+ influx occurring with each action potential, in the presence of diminished outward Na+ pumping due to digitalis, leads to the increased intracellular Na+ concentration proposed to promote increased intracellular Ca2+ stores, either through enhanced Ca2+ entry, reduced Ca2+ efflux, or both. These effects are thought to be mediated via Na+-Ca2+ exchange. At rapid heart rates, Na+ influx in the presence or absence of digitalis may be sufficiently great to approach the maximum inotropically effective intracellular Na+. This is consistent with the diminished effects of cardiac glycosides at high contraction frequencies.
Studies with spontaneously contracting, isolated, and cultured ventricular cells have demonstrated direct correlations between ouabain-induced enhancement of the contractile state, inhibition of Na+ and K+ transport, and increased cellular content of both Na+ and Ca2+ (7). These data support the hypothesis that inhibition of the Na+ pump by digitalis leads to a positive inotropic response and are consistent with modulation of Ca2+ content by Na+ via the Na+-Ca2+ exchange carrier mechanism (Fig. 25-1). A similar effect can be achieved by decreasing
extracellular [K+], which can produce equivalent positive inotropic effects accompanied by inhibition of Na+ and K+ transport and an increase in intracellular [Na+] and intracellular calcium activity, [Ca2+]i, via Na+-Ca2+ exchange (19). Direct evidence for an increase in [Ca2+]i following digitalis comes from studies using the Ca2+-activated photoprotein aequorin, as well as Ca2+-specific fluorescent probes such as fura-2 and indo-1. During the inotropic effect of digitalis, there is a close association between the increase in peak systolic aequorin luminescence, which is directly related to intracellular [Ca2+] over the relevant range, and the increase in twitch force.
extracellular [K+], which can produce equivalent positive inotropic effects accompanied by inhibition of Na+ and K+ transport and an increase in intracellular [Na+] and intracellular calcium activity, [Ca2+]i, via Na+-Ca2+ exchange (19). Direct evidence for an increase in [Ca2+]i following digitalis comes from studies using the Ca2+-activated photoprotein aequorin, as well as Ca2+-specific fluorescent probes such as fura-2 and indo-1. During the inotropic effect of digitalis, there is a close association between the increase in peak systolic aequorin luminescence, which is directly related to intracellular [Ca2+] over the relevant range, and the increase in twitch force.
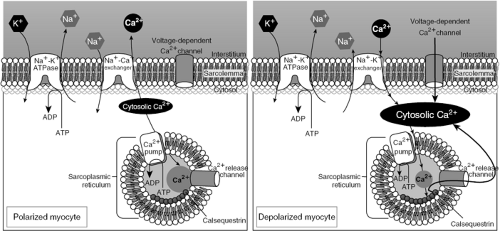 Figure 25-1 The figure illustrates the flux of sodium (Na+), potassium (K+), and calcium (Ca2+) across the plasmalemmal membrane and sarcoplasmic reticular membrane, during diastole (left panel) and systole (right panel). Digitalis glycosides such as digoxin bind to the Na+-K+-ATPase, and decrease the flux of potassium into, and sodium out of, the cell, resulting in increased levels of intracellular Na+. This increase in intracellular Na+ results, among other effects, in increased Na+/Ca2+ exchange, a subsequent rise in intracellular Ca2+, including SR calcium content, which results in a modest increase in contractile force during each cardiac cycle. Other mechanisms of action of the cardiac glycosides at drug levels achieved using clinically relevant dosing algorithms have been postulated, including direct effects on ryanodine receptors. |
The immediate positively inotropic effect of cardiac glycosides, measured either in the intact heart in a conscious dog model or in isometrically contracting papillary muscle strips, is achieved with remarkable energy transfer efficiency and little oxygen wasting (20). When compared with β-adrenergic agonists or a cyclic adenosine monophosphate phosphodiesterase inhibitor, ouabain caused no significant change, for the same degree of tension development, in the tension-time integral per unit initial heat, an index of the economy of isometric contraction in an isolated muscle preparation. Interestingly, Peng et al. (21) have shown that ouabain-induced net influx of Ca2+ induces c-fos and other early response gene transcription. Huang et al. (22,23) also have demonstrated that sodium pump inhibition can lead to cardiac myocyte hypertrophy in vitro, accompanied by a characteristic phenotypic shift to a fetal gene program in these cells. Whether these observations in cell culture preparations are relevant to the clinical pharmacology of these drugs in humans is not known.
Electrophysiological Effects
As with the positive inotropic effect of these drugs, the major effect on cardiac rhythm of digitalis preparations is believed to be due to inhibition of the sodium pump. The 80- to 90-mV transmembrane resting potential of cardiac cells is maintained by Na+ and K+ gradients (particularly the latter), which in turn are dependent on the integrity of the active Na,K pump mechanism. There is general agreement that inhibition of Na,K-ATPase underlies the direct toxic effects of digitalis preparations on cardiac rhythm, and thus represents an extension of the drugs’ therapeutic effects. Cells in various parts of the heart show differing sensitivities to digitalis, and both direct and neurally mediated effects must be dissected before conclusions can be drawn about the mechanisms involved. For example, digoxin does not produce significant changes in atrial effective or functional refractory periods in denervated hearts, nor does it change sinus node cycle length. These findings point to the importance of the autonomic nervous system in the regulation of automaticity and conduction by digitalis. Indeed, at the lower concentrations associated with therapeutic levels of digoxin, the drugs decrease automaticity and increase maximum diastolic potential (effects that can be blocked by atropine), whereas higher concentrations decrease diastolic potentials and increase automaticity.
Similarly, the toxic arrhythmogenic effect of the cardiac glycosides is due to a combination of direct effects on the myocardium and neurally mediated increases in autonomic activity. During exposure to high concentrations of digitalis, isolated myocardial preparations demonstrate small, unstimulated depolarizations and contractions following action potentials. These oscillatory events coincide with the development of digitalis-toxic arrhythmias in intact animals exposed to similar levels of digitalis. Both systolic and diastolic [Ca2+]i increase during digitalis-induced arrhythmias, increases that were first inferred from changes in tension, leading to the idea that intracellular calcium overload contributes to the observed arrhythmogenic effects. These occur presumably because [Ca2+]i increases progressively until the sarcoplasmic reticulum (the major intracellular organelle responsible for Ca2+ sequestration) is no longer capable of retaining all the Ca2+ taken up with each depolarization. Spontaneous cycles of Ca release and reuptake then ensue, resulting in after depolarizations and after contractions. The after-depolarization is the result of a Ca2+-activated transient outward current and is thought to be the macroscopic manifestation of Ca-activated nonspecific cation channels, plus Na-Ca exchange current (ITO). This formulation has been confirmed by Hancox and Levi in rabbit atrioventricular nodal tissue (24). Interestingly, mice that lack a functioning endothelial nitric oxide synthase (eNOS) isoform gene are more susceptible to digitalis-induced increases in ITO and after-depolarizations (Han, Kubota, and Kelly, unpublished observations [25]). This implicates local, cardiac myocyte-derived nitric oxide (NO) in the regulation of transmembrane Ca2+ currents in digitalis-treated animals (25,26,27).
Neurally Mediated Actions of Cardiac Glycosides
It is important to understand the pathophysiology of heart failure and the role of digitalis glycosides in modifying the abnormal autonomic nervous system activity characteristic of advanced heart failure, including altered baroreflex activity. The responsiveness of the baroreflex system is diminished in congestive heart failure. Because stimulation of this reflex normally inhibits sympathetic outflow, the result of diminished baroreflex activity in these patients is increased sympathetic nerve activity, as well as increased vasopressin.
Mason et al. (28) observed that intravenous ouabain increased mean arterial pressure, forearm vascular resistance, and venous tone in normal human subjects, probably due to direct but transient effects on vascular smooth muscle. In contrast, patients with heart failure responded with a decline in heart rate and other effects that were consistent with enhanced baroreflex responsiveness. Direct effects of cardiac glycosides on carotid baroreflex responsiveness to changes in carotid sinus pressure have been reported in isolated baroreceptor preparations from animals with experimentally induced heart failure. Ferguson et al. (29) demonstrated in patients with moderate to severe heart failure that infusion of deslanoside increased forearm blood flow and cardiac index and decreased heart rate, concomitant with a marked decrease in skeletal muscle sympathetic nerve activity measured as an indicator of
centrally mediated sympathetic nervous system activity. Indeed, the neurohormonal effects are quite broad; in addition to attenuation of carotid sinus baroreceptor discharge sensitivity, digoxin has vasomimetic and sympathoinhibitory effects and effects that decrease serum norepinephrine, plasma renin activity, and possibly aldosterone (30). Based on these and other observations, reduced neurohumoral activation may be the most significant mechanism contributing to the efficacy of cardiac glycosides in the treatment of patients with heart failure, and may occur at blood levels of these drugs that are below those necessary to achieve a direct inotropic effect (31).
centrally mediated sympathetic nervous system activity. Indeed, the neurohormonal effects are quite broad; in addition to attenuation of carotid sinus baroreceptor discharge sensitivity, digoxin has vasomimetic and sympathoinhibitory effects and effects that decrease serum norepinephrine, plasma renin activity, and possibly aldosterone (30). Based on these and other observations, reduced neurohumoral activation may be the most significant mechanism contributing to the efficacy of cardiac glycosides in the treatment of patients with heart failure, and may occur at blood levels of these drugs that are below those necessary to achieve a direct inotropic effect (31).
Clinical Pharmacology
Pharmacokinetics
Although a number of cardiac glycoside preparations remain available, digoxin is the most commonly prescribed; only its pharmacology will be described here in detail. The reader should refer to the first edition of this text for a description of the pharmacology of other cardiac glycosides that remain in clinical use.
Digoxin is excreted exponentially, with an elimination half-life of 36 to 48 hours in patients with normal renal function, resulting in the loss of about one-third of body stores daily. The drug is excreted for the most part unchanged, although some patients excrete detectable quantities of the inactive metabolite dihydrodigoxin, which arises through bacterial biotransformation in the gut lumen. Renal excretion of digoxin is proportional to the glomerular filtration rate (and, hence, to creatinine clearance). In patients with prerenal azotemia, digoxin clearance may correlate more closely with urea clearance, suggesting that the drug may undergo some degree of tubular reabsorption.
With daily maintenance therapy, a steady state is reached when daily losses are matched by daily intake. For patients not previously given digoxin, institution of daily maintenance therapy without a loading dose results in development of steady-state plateau concentrations after four to five half-lives, or about 7 days, in subjects with normal renal function. If the half-life of the drug is prolonged, the length of time before a steady state is reached on a daily maintenance dose would also be prolonged proportionately. Because of the high degree of tissue binding of digoxin (i.e., a large volume of distribution, averaging 4 to 7 L/kg), the drug is not effectively removed from the body by dialysis. Serum digoxin levels and pharmacokinetics are essentially the same before and after the loss of large amounts of adipose tissue in massively obese subjects, suggesting that a patient’s estimated lean body mass should be used in the calculation for maintenance dosing. Finally, the steady-state volume of distribution of digoxin (Vdss) is decreased in chronic renal failure, and therefore loading doses of digoxin as well as maintenance doses should be decreased in these patients (32).
In patients with advanced heart failure, initiation of therapy with vasodilators tends to increase renal digoxin clearance (probably by increasing cardiac output) and may necessitate adjustment of the maintenance digoxin dosage. Infants and children absorb and excrete digoxin in much the same way as adults do, although recent evidence suggests that secretion at the renal tubular level may be quantitatively more important in children before puberty. Digoxin doses in neonates and infants are substantially larger than those in adults when adjusted for weight or per square meter of body surface area. These higher doses result in relatively higher serum digoxin concentrations, which are generally well-tolerated. Digoxin does cross the placenta and fetal umbilical cord venous blood levels of the drug are similar to maternal blood levels. There is no contraindication for use of digoxin during pregnancy or during lactation.
Numerous studies over several decades have documented incomplete absorption of digoxin following oral administration, with the bioavailability of currently marketed tablet preparations averaging about 65% to 75%. Individual patient variation, interactions with other concurrently administered drugs, and the characteristics of the digoxin preparation ingested are all known to affect the drug’s bioavailability. Patients with malabsorption syndromes often absorb digoxin poorly and erratically. However, patients with pancreatic insufficiency, despite steatorrhea, appear to absorb the drug normally. Administration of digoxin after meals is likely to delay absorption, thus diminishing peak serum levels, but absolute bioavailability is not affected to any noteworthy degree. Absorption of digoxin tends to be reduced by drugs that increase motility, particularly if a particular formulation releases the active drug slowly. In addition, nonabsorbable substances such as cholestyramine and nonabsorbable antacids, when taken concurrently with digoxin, can interfere with gastrointestinal absorption of digoxin.
Digoxin Dosing Schedules
There is usually no need to treat patients with a loading dose of digoxin except in the setting of certain supraventricular arrhythmias when other drugs useful in treating these arrhythmias are contraindicated or have not been effective. This is due to the narrow therapeutic window of cardiac glycosides, which often makes it difficult to judge accurately an effective loading dose of digoxin that also will minimize the risk of toxicity. Often the patients who would benefit most from the addition of a cardiac glycoside are also those at greatest risk of exhibiting toxic effects of these drugs. Nevertheless, if a loading dose is to be given (assuming a bioavailability of 75% to 80% for most digoxin tablet formulations), between 0.9 and 1.8 mg given in divided doses over 24 hours will result in plasma levels that approximate those achieved by either 0.25 or 0.50 mg of digoxin, respectively, given daily for about a week in patients with normal renal function. Lean body mass should be used in the calculation of both loading and maintenance digoxin doses. In adults, intravenous loading doses of 0.50 to 0.75 mg per 45 kg (i.e., 100 pounds) of body weight, in divided doses over 24 hours, are unlikely to cause toxicity and can be supplemented as required by the patient’s clinical condition. The loading dose of digoxin should be reduced in
patients with renal insufficiency due to a reduction in the drug’s volume of distribution in this condition, as previously noted.
patients with renal insufficiency due to a reduction in the drug’s volume of distribution in this condition, as previously noted.
The calculation of the regular maintenance dose is determined by the drug’s clearance rate from the body, which, in the case of digoxin, is closely related to the rate of clearance of creatinine. A reasonable approximation of the daily percentage loss of digoxin is given by:
The daily maintenance dose (33,34) is calculated by multiplying the percentage daily loss times the loading dose given that resulted in effective therapeutic drug levels (adjusting for reduced bioavailability if the maintenance dose is to be given orally, but the loading dose was given intravenously).
Drug Interactions
Concomitant drug administration may directly alter the pharmacokinetics of digitalis preparations, or indirectly alter their action on the heart by pharmacodynamic interactions (Table 25-1). Quinidine reduces both the renal and nonrenal elimination of digoxin and decreases its volume of distribution. The net result is an increase in serum digoxin concentration that averages twofold in patients given conventional doses of quinidine. Unfortunately, individual responses to quinidine may vary and close surveillance of clinical status and the serum digoxin concentration is warranted. Procainamide and disopyramide do not appear to alter serum digoxin levels, but verapamil does increase the serum digoxin concentration by an average of 35%, again by decreasing digoxin’s volume of distribution and clearance rate. Nifedipine and diltiazem have no reproducible effect on digoxin clearance. Both short-term and long-term amiodarone administration has been found to increase the steady-state digoxin concentration, and maintenance doses of digoxin should be decreased by half (or more). Newly introduced drugs will require close surveillance for interactions with cardiac glycosides (35,36). Furthermore, over-the-counter drugs and nutraceuticals (37,38) also may interact with digoxin clearance.
Other examples of pharmacodynamic interactions include concomitantly administered diuretic agents that may increase the incidence of digitalis toxicity both by decreasing the glomerular filtration rate due to volume depletion and by inducing a variety of electrolyte disturbances, including hypokalemia, hypomagnesemia, and (for thiazide diuretics) hypercalcemia. Also, concurrent administration of some antiarrhythmic agents may increase the possibility of proarrhythmic events, an outcome that is often unpredictable in an individual patient.
Several anesthetic agents, such as cyclopropane and succinylcholine, may demonstrate additive or synergistic proarrhythmic effects when coadministered with digitalis. Experimental studies demonstrate that catecholamine-induced increases in ventricular automaticity add to the arrhythmogenic effects of digitalis. Therefore, it is reasonable
to assume that sympathomimetic agents increase the likelihood of enhanced automaticity of atrial and ventricular tissue in patients receiving digitalis.
to assume that sympathomimetic agents increase the likelihood of enhanced automaticity of atrial and ventricular tissue in patients receiving digitalis.
Table 25-1 Pharmacokinetic and Pharmacodynamic Interactions with Digoxin | |||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||
Cardiac Glycosides and Heart Failure
Despite the fact that digitalis has been used for the treatment of symptoms of congestive heart failure for over 200 years, it was not until early in the twentieth century that a series of clinical observations led to the recognition that these drugs could be useful in patients with normal sinus rhythm.
Digitalis in Patients with Congestive Heart Failure in Sinus Rhythm: Early Clinical Trials
Clinical studies appeared in the 1960s and 1970s that questioned Christian’s assertion that patients in sinus rhythm with heart failure clearly benefited from digitalis. These studies suggested that many patients receiving chronic maintenance doses of digoxin did not benefit from the drug (42,43). A number of small, double-blinded, placebo-controlled trials involving 22 to 30 patients appeared in the period from 1980 to 1992, yielding conflicting results (44,45,46).
Changes in quantitative measures of contractile function in these and other studies were variably correlated with symptoms or exercise performance, where such correlations were sought. In general, patients with more severe contractile dysfunction demonstrated the most pronounced beneficial response to digoxin. This was particularly evident in a study by Gheorghiade et al. (47), in which marked improvements in mean cardiac output (+48%), left ventricular filling pressure (-36%), and ejection fraction (+8%) were observed in the subset of patients who remained in overt failure after diuretic and vasodilator treatment, whereas no significant further improvement was observed in the subset of patients who were relatively well-compensated at the completion of the diuretic-vasodilator treatment period.
Several larger, multicenter trials also appeared at the beginning of the 1990s. The digoxin and xamoterol study examined 433 patients (the majority had New York Heart Association [NYHA] Class I or II failure). Xamoterol is a partial β1-selective adrenergic agonist with some antagonist activity (48). Patients were randomized to placebo, xamoterol, or digoxin in a 1:2:1 ratio; diuretics were continued but were limited to no more than 80 mg per day of furosemide or equivalent. Three hundred patients completed the double-blind phase with valid exercise tests at entry and at 3 months. Digoxin did not improve exercise duration or work done on a bicycle ergometer. However, although both digoxin and xamoterol demonstrated a significant improvement in symptoms compared with placebo, xamoterol was subsequently shown to cause excess mortality in patients with heart failure.
The captopril-digoxin trial compared captopril, digoxin, and placebo during maintenance diuretic therapy in 196 patients, 85% of whom were judged to be in NYHA Class I or II (49). Patients who did not tolerate withdrawal from digoxin were not randomized, thus excluding the subset of patients who were presumably the most likely to demonstrate benefit from digoxin. Two major conclusions emerged from this trial: digoxin (but not captopril) significantly improved the left ventricular ejection fraction, and digoxin and captopril were both similarly effective in reducing morbidity (by about half) compared with placebo in terms of increased diuretic requirements and hospitalization and emergency room visits.
The milrinone-digoxin trial randomly assigned 230 patients in sinus rhythm with moderately severe heart failure to treatment with digoxin, oral milrinone, both drugs, or placebo added to baseline diuretic therapy (50). After 3 months, digoxin improved exercise tolerance by 14% (p <0.05) compared with placebo. There was a marked benefit of digoxin over placebo or milrinone as judged by the frequency of decompensation within the initial 2 weeks and at 3 months. There also was a significantly lower incidence of ventricular ectopy in the digoxin-treated group, compared with those patients randomized to receive milrinone. Oral milrinone was subsequently documented to increase mortality in patients with heart failure.
Digoxin Withdrawal Trials
The PROVED (Prospective Randomized Study of Ventricular Failure and Efficacy of Digoxin) (51) and RADIANCE (Randomized Assessment of Digoxin on Inhibition of Angiotensin-Converting Enzyme) (52) trials were two prospective, multicenter, placebo-controlled trials that examined the effects of withdrawal of digoxin in patients with stable, mild to moderate heart failure (i.e., NYHA Class II and III) and systolic ventricular dysfunction (i.e., a left ventricular ejection fraction of ≤0.35%). All patients studied were in sinus rhythm. The target serum digoxin concentration in both studies during the baseline run-in phase was 0.9 to 2.0 ng/mL, achieved with an average digoxin dose of 0.38 mg per day. Patients in the RADIANCE trial also received concurrent therapy with an angiotensin-converting enzyme (ACE) inhibitor. When patients were randomly assigned to either continue active digoxin therapy or to withdraw from active therapy and receive a matching placebo, 40% of patients in PROVED and 28% of patients in RADIANCE who received placebo noted a significant worsening of heart failure symptoms compared with 20% and 6%, respectively, in patients who continued to receive active drug. This absolute risk reduction of 20% in digoxin-treated patients constituted a substantial treatment effect. Maximal treadmill exercise tolerance also declined significantly in patients withdrawn from digoxin in both trials, despite continuation of other medical therapies for heart failure, notably ACE inhibitors in RADIANCE (52). In addition, the benefits of digoxin (compared to withdrawal) were seen over the range of serum levels of drug, somewhat in contrast to the conclusions derived from the post hoc analysis of the Digoxin Investigation Group (DIG) trial (53) (see The Digoxin Investigation Group Trial later). Overall, however, none of the withdrawal trials had the statistical power to detect an effect of digoxin therapy on the survival of patients with heart failure.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


