Fig. 3.1
Hypothesis of dysregulated inflammatory/stress/metabolic interaction mechanisms in metabolic syndrome. MS metabolic syndrome, CRP C-reactive protein, IL-6 interleukin-6, NA noradrenaline, eHsp72 72 kDa extracellular heat shock protein
Local Inflammatory Activity and Its Dysregulation in Response to Stress Mediators
The systemic dysregulation between inflammatory and stress biomarkers in MetS may be just a consequence of the altered inflammatory and stress responses locally. A local inflammation is observed in the adipose tissue, liver, and skeletal muscle in the pathogenesis of MetS and T2D, but its role in obesity-related metabolic disorders remains to be well determined [7]. It has been increasingly recognized that an imbalance of pro-inflammatory and anti-inflammatory adipokines contributes to the development of obesity-linked disorders. Dysregulation of anti-inflammatory adipokines caused by fat accumulation also participates in local or systemic inflammatory responses, thereby leading to the initiation or progression of metabolic and CVD [25]. Thus, there has been described a dysregulation in the synthesis and release of adipocytokines by adipocytes or by infiltrated macrophages in the adipose tissue of obese subjects: elevated secretion of the pro-inflammatory adipocytokines (TNF-α, IL-6, haptoglobin, leptin, resistin) and reduced secretion of the anti-inflammatory ones (adiponectin). Infiltration of macrophages into tissues is an important source of circulating inflammatory molecules [26]. These pro-inflammatory cytokines (including TNF-α, IL-6, and IL-1β) produced by infiltrated macrophages act in an autocrine and paracrine manner to promote IR by interfering with insulin signaling in peripheral tissues through activation of the c-JUN N-terminal kinase (JNK) and nuclear factor-kappa B (NF-kB) pathways [27]. Therefore, adipose tissue appears to play a central role in the induction of inflammation, and over-nutrition leads to changes in its cellular composition and production of pro-inflammatory cytokines and chemokines [7].
As reviewed by Esser and coworkers [7], macrophages can be classified into two distinct subtypes: the “classically activated macrophages,” a pro-inflammatory phenotype termed M1 macrophages which release pro-inflammatory cytokines such as IL-1β, IL-6, TNF-α, and nitric oxide (NO), and the “alternatively activated macrophages,” an anti-inflammatory phenotype termed M2 macrophages which release anti-inflammatory cytokines such as IL-10 [28]. While well established in mice [29, 30], the existence of M1 and M2 macrophage subsets has not been confirmed in human adipose tissue, where macrophages seem to be a mix of the M1 and M2 phenotypes [31]. Nonobese, physically active, and healthy dieted people maintain an “anti-inflammatory phenotype of adipose tissue” (small adipocyte size and the presence of M2 macrophages). Increased visceral fat induced by a positive energy balance and physical inactivity provokes “inflamed adipose tissue” characterized by predominant infiltrated M1 macrophages [32]. In addition, obesity can cause a phenotypic switch from the M2 to M1 phenotype, correlating with IR in both mice and humans [29–31]. Direct and paracrine signals issued from M1 macrophages can impair insulin signaling and adipogenesis in adipocytes, whereas M2 macrophages seem to protect against obesity-induced IR [28]. In addition, there is evidence that both TLR2 and TLR4 (toll-like receptors involved in the innate and inflammatory responses of immune cells, particularly macrophages) can recognize fatty acids that contribute to the release of pro-inflammatory cytokines by macrophages [33].
In obese Zucker rats, it has also been observed that peritoneal macrophages (not infiltrated in adipose tissue) exhibit a local dysregulation with respect to the constitutive or spontaneous release of pro-inflammatory cytokines, which could also be contributing to the low-grade systemic pro-inflammatory state in MetS and thus affecting the development of CVD [3, 34].
Also, the peritoneal macrophages not infiltrated in the adipose tissue show impaired release of such pro-inflammatory cytokines as IL-1β, INF-γ [34], IL-6, and TNF-α [3] in response to an antigen (lipopolysaccharide) stimulus, thus potentially increasing susceptibility to infection. This functional defect may be one of the causes of the increased susceptibility to candidiasis observed in obese individuals [35]. Indeed, the increased candidicide activity of macrophages of the obese animals could be considered to be an adaptation to this phenomenon [36]. As indicated previously, these cells also exhibit altered release of inflammatory cytokines in response to “stress messengers” or “endogenous danger signals” such as NA and Hsp72, even to animals with MetS changing their stimulatory or inhibitory effects on the release of inflammatory cytokines, particularly of IL-6 and IL-1β [20, 24]. It is therefore plausible to conclude that, in addition to systemic dysregulation in MetS, there appears local dysregulation of the innate and inflammatory responses, which not only involves infiltrated macrophages but also non-infiltrated macrophages such as the peritoneal macrophages as is manifested both in fewer of these cells in the peritoneal exudate (which may be due to increased infiltration in adipose tissue) and in impaired functional capacity [3, 20, 24, 34]. All of this serves to confirm the altered innate and inflammatory immune response capacity mediated by macrophages in MetS, corroborating the hypothesis of dysregulation in the neuroendocrine-immune response that can arise in this pathology.
Altered Response to Exercise-Induced Stress in the Metabolic Syndrome
It is now clear that obesity, T2D, and CVD share an environment characterized by IR and inflammation [27]. Therefore, removal of the inflammatory mediator signals is a more than promising strategy for the management of MetS. The potential anti-inflammatory effects of exercise have been strongly proposed as being an essential exercise-induced mechanism for improving diabetic status and insulin sensitivity by controlling the low-grade systemic inflammation associated with MetS [4, 32, 34, 37]. It has been explained above, as proposed by Gleeson and coworkers, that while a healthy diet together with exercise maintains the anti-inflammatory phenotype of adipose tissue (with predominant M2 anti-inflammatory macrophages), inactivity and an excess of energy balance lead to pro-inflammatory M1 macrophages predominating in the “inflamed adipose tissue.” In this way, regular exercise decreases the risk of chronic metabolic and CVD in part because exercise exerts anti-inflammatory effects that may be mediated by both a reduction in the visceral fat mass and the induction of an anti-inflammatory environment [32]. In addition, exercise training not only inhibits inflammation in adipose tissue via suppression of macrophage infiltration but also through a phenotypic switch from M1 to M2 macrophages [38].
Nevertheless, it is clearly accepted that exercise is a form of stress and that the relation between exercise, stress, and inflammation is a good model of neuroendocrine interactions. Exercise stimulates the innate immune responses, and its effects on the inflammatory response are primarily mediated through the hypothalamic-pituitary-adrenal (HPA) axis and the SNS. A well-controlled and regulated stimulation of the innate and/or inflammatory mechanisms during exercise can help to prevent infection, but over-stimulation of the inflammatory response could also be harmful for people with inflammatory diseases, as it is the case in MetS and cardiovascular-associated disorders [3]. Thus, the anti-inflammatory hypothesis of exercise and its physiological implications needs to be clarified, particularly in the management of pathologies with inflammatory dysregulation, such as MetS. Today, it is still not formally proven whether or not an induced anti-inflammatory effect of exercise in healthy people (with an optimal neuroendocrine and inflammatory feedback) is good for the optimal regulation of homeostasis. The anti-inflammatory effects of exercise should be mainly (or only) beneficial for people with an unhealthily high inflammatory status. Further studies to define the duration and intensities of exercise programs that restore anti-inflammatory responses and the optimal cytokine-HPA axis feedback circuit, avoiding unhealthy pro-inflammatory and stress responses, are clearly necessary. For example, an inappropriate intensity of regular exercise performed by animals with MetS presenting a dysregulation in the inflammatory/stress feedback mechanisms (mediated by IL-6 and NA) can worsen this dysregulation still more by increasing circulating levels of NA, IL-6, and glucose [3] and by increasing the tissue expression and circulating levels of TNF-α, all together with a higher innate (phagocytic and microbicidal capacity) response of peritoneal macrophages [3]. These effects seem to reflect that intense regular exercise, while inducing a greater protection against infection, could exacerbate some aspects of inflammatory conditions, and intensity and duration of exercise inducing anti-inflammatory effects could compromise the immune system against pathogen challenges.
Altered response to exercise-induced stress in MetS is also manifest after single bouts of exercise. Thus, the regulation by exercise of the altered inflammatory and stress status in MetS depends on the basal set point of each individual, being anti-inflammatory mainly (or only) during a high inflammatory status. Thus, acute exercise only decreased the systemic level of IL-6, NA (also dopamine), and glucose in obese rats with the highest basal levels of these biomarkers [3].
In sum, it seems that both the pro-inflammatory and the anti-inflammatory effects of exercise may cause an additional cost (side effect) in chronic low-grade inflammatory diseases such as MetS, exacerbating the inflammatory condition (if pro-inflammatory) or increasing the susceptibility to infections (if anti-inflammatory). A good “bioregulation” of adequate intensities and durations of exercise needs to achieve a decrease in unhealthy inflammatory biomarkers together with optimal phagocytic and microbicidal activities, and clearly further studies are needed in this direction.
Molecular Mechanisms Implicated in Cardiometabolic Risk
The aim of this section is to highlight prominent molecular signals and pathways in T2D that have been linked to the development of cardiovascular disease in humans. A particular emphasis is placed on processes that have been implicated in both the pathogenesis of diabetes itself and its vascular and cardiac complications, since it is increasingly clear that these conditions often coexist as a pathologic continuum collectively referred to as “cardiometabolic disease.” This chapter section is arranged into three essential parts: (1) circulating factors that have been linked to the development of diabetes and cardiovascular disease, (2) prominent molecular mechanisms that lead to vascular dysfunction and disease in diabetes, and (3) links between diabetes and the development of cardiomyopathy (heart failure).
Circulating Markers of Cardiovascular Disease in Diabetes
This part highlights three major biomarkers of diabetes that are both predictive of cardiovascular complications in epidemiological studies and mechanistically linked to the pathogenesis of diabetes and cardiovascular disease in basic and/or clinical studies.
Glucose Homeostasis Disruption
Diabetes mellitus encompasses multiple pathologies with distinct etiologies. Type 1 diabetes (T1D) is an autoimmune disease that results from a loss of pancreatic β-cells, thereby eliminating insulin production and systemic response to hyperglycemia. T2D is characterized in its early stage by impaired uptake of glucose by peripheral tissues in response to insulin, a phenomenon known as IR. Pancreatic β-cells are initially able to compensate for the resulting hyperglycemia by increasing insulin production and secretion, but this results in progressive cell hypertrophy and stress that ultimately leads to β-cell loss. Thus, the late stage of type 2 disease resembles T1D. Overall, diabetes mellitus is characterized by the inability to maintain glucose homeostasis with the consequent extended periods of hyperglycemia and also the dangerous appearance of hypoglycemia episodes.
Multiple epidemiological studies indicate that diabetic patients have an increased incidence of cardiovascular diseases compared with nondiabetic subjects. Extended daily periods of hyperglycemia are a major reason for this elevated cardiovascular risk [39]. Chronic and postprandial hyperglycemia promotes biochemical reactions leading to the glycation of proteins, lipids, or nucleic acids in peripheral tissues, generating a variety of compounds collectively known as advanced glycation end products (AGEs). The kidney is the major organ responsible for AGE clearance, which could explain the association of increased levels of circulating AGEs with renal dysfunction in diabetics. AGEs can act extracellularly or bind to receptors that signal intracellularly, such us AGEs receptor (RAGEs), to trigger several pathogenic effects on the vasculature and heart discussed further in subsequent sections.
It is important to mention that in addition to hyperglycemia, it has been shown that glucose fluctuations (hyperglycemic peaks and hypoglycemic valleys) trigger inflammatory responses and oxidative stress leading to vascular endothelial dysfunction and increased cardiovascular risk [40]. Therefore, a full understanding of the pathogenic effects elicited by poor glucose control requires well-controlled clinical studies employing continuous glucose measurement systems addressing glucose variability in the development of vascular complications.
Adipokine Imbalance
Obesity accounts for more than 80 % of the risk of developing T2D. Physical inactivity and an increased energy intake due to a diet rich in simple carbohydrates and saturated fat are related to the growing incidence of overweight and obesity around the world. The World Health Organization reported that in 2008 more than 1.4 billion adults were either overweight or already obese. Obesity is characterized by excessive fat accumulation in adipose tissue as well as ectopic lipid accumulation in organs such us liver, skeletal muscle, and cardiac muscle. Adipose tissue is not only the major tissue for lipid storage but also plays a critical role as an endocrine organ. The peptide hormones secreted by adipose tissue are known as adipokines or adipocytokines. In the last decades a growing number of studies demonstrated a clear relationship between adipokine levels and cardiovascular complications [41]. Pro-inflammatory cytokines have been discussed in a previous section of this chapter, therefore discussion will be limited specifically to the effects of adipokines.
Adipokine production and secretion responds to changes in adipose tissue mass, stress, and inflammatory states that could, through multiple mechanisms, influence cardiovascular disease risk. Leptin was the first adipokine discovered in 1994. This hormone is a major regulator of energy balance by direct actions on hypothalamic neurons that regulate food intake and energy expenditure. Circulating leptin levels are directly correlated with adipose tissue mass. Thus, obesity-related T2D is characterized by hyperleptinemia and leptin resistance. It has been reported that chronic hyperleptinemia may promote atherosclerosis by stimulating monocyte migration, increasing production of reactive oxygen species (ROS) in endothelial cells, and promoting vascular inflammation and thrombosis [42]. Therefore, interventions aimed at reducing circulating leptin levels and/or improving leptin sensitivity could have a positive impact on cardiovascular risk. Indeed, lowering circulating leptin levels may be one of the positive effects of weight loss after gastric bypass surgery.
Adiponectin is another adipokine primarily expressed and secreted by mature adipocytes. Its main functions are the stimulation of fatty acid oxidation and reduction of hepatic gluconeogenesis and plasma glucose levels. In contrast to leptin, adiponectin has anti-inflammatory and anti-atherogenic properties. Unfortunately, circulating adiponectin levels are significantly reduced in obesity-related T2D, which may result from elevations of pro-inflammatory cytokines, such as TNF-α, known to suppress adipocyte adiponectin expression. Thus, interventions that increase plasma levels of adiponectin could be beneficial to prevent cardiovascular risk or ameliorate cardiovascular complications. For example, adipose tissue reduction by bariatric surgery leads to increased levels of circulating adiponectin and potently reduces cardiovascular risk [43]. Drug treatments such as peroxisome proliferator-activated receptor gamma (PPAR-γ) agonists (e.g., pioglitazone) and angiotensin-converting enzyme inhibitors have also been successful raising circulating adiponectin levels, with well-known cardioprotective effects.
Other adipokines have also been studied, but less extensively, to assess their potential role in cardiovascular disease; this is the case of resistin and omentin-1. Circulating levels of resistin are elevated in obese and T2D subjects. Resistin is not secreted by adipocytes but is expressed in macrophages, which could explain its role in inflammation. Regarding cardiovascular risk, resistin has been related to the development of endothelial dysfunction, thrombosis, angiogenesis, and smooth muscle dysfunction [44]. Omentin-1 elicits anti-inflammatory and insulin-sensitizing effects, while also promoting vasodilation, but in a similar manner to adiponectin, omentin-1 expression is downregulated in obesity and T2D [45].
Diabetic Dyslipidemia
While the damaging effect of hyperglycemia on the cardiovascular system is well established, improving glucose control only modestly reduces cardiovascular risk in diabetic populations [46]. It is becoming increasingly recognized that aggressive management of diabetic dyslipidemia is also of critical importance. Chronic elevations in circulating free fatty acids in obesity/diabetes increase hepatic production and release of triglyceride-rich VLDL. This promotes a characteristic lipid “triad” of elevated serum triglycerides, small-dense low-density lipoprotein cholesterol (sdLDL-C), and reduced HDL-C, often with no change in total LDL [47]. Elevations of circulating free fatty acids are also implicated in the pathogenesis of IR; thus targeting this early lipid abnormality (e.g., with fibrates or thiazolidinediones) may substantially reduce cardiometabolic risk in patients with diabetes and MetS.
Mechanistic Links Between Diabetes and Vascular Dysfunction
Following the general order of topics from the previous section, a summary of the mechanistic roles of these biomarker/systems in the pathogenesis of vascular dysfunction/disease in diabetics is detailed.
Hyperglycemia Increases Production of Reactive Oxygen Species and Advanced Glycation End Products
As already mentioned, the uncontrolled and extended daily periods of hyperglycemia characteristic of diabetes are mediators of the increased cardiovascular risk observed in diabetic patients. Regarding vascular complications, endothelial dysfunction is an early event in the etiology of diabetes that leads to atherosclerosis in the long term. Hyperglycemia leads to mitochondrial dysfunction and increased ROS production in endothelial cells, compounded by elevated flux through the pentose phosphate pathway (exacerbating ROS production) and the formation of AGEs. The increase in ROS impairs angiogenesis and induces a metabolic switch reducing glycolysis and activating fatty acid oxidation, both of which may lead to vascular dysfunction. AGEs alter the extracellular matrix leading to vascular complications. AGEs act intracellularly through the activation of the receptor for AGEs to promote vascular inflammation and activate signaling cascades that reduce NO production and increase ROS production mediated by nicotinamide adenine dinucleotide phosphate (NADPH) oxidase [48]. AGEs also activate mitogen-activated protein kinases (MAPK), which along with ROS are able to activate the nuclear transcription factor NF-κB, which triggers the transcription of several pro-inflammatory cytokines, intracellular adhesion molecules, and the vascular endothelial growth factor (VEGF) [49]. Hyperglycemia also impairs the ability of endothelial nitric oxide synthase (eNOS) to produce nitric oxide (NO) by depleting arginine (substrate needed for the NO formation), which “uncouples” the enzyme leading to superoxide production. This further reduces NO bioavailability and increases production of peroxynitrite, which elicits toxic effects by oxidizing and nitrosylating cellular proteins such as those involved in oxidative phosphorylation in mitochondria, further promoting endothelial dysfunction.
Imbalance of Adipokine Signals
As mentioned above, obesity-related T2D patients are characterized by high levels of circulating leptin. Hyperleptinemia has been identified as an independent risk factor, mediating endothelial dysfunction and neointimal hyperplasia leading to atherosclerotic coronary artery disease and arterial hypertension [42]. The molecular mechanisms implicated in leptin actions at the cellular level are mediated through different leptin receptors. Leptin receptor signaling interacts with several different signaling pathways, mainly Janus kinase/signal transducers and activators of transcription (JAK/STAT) and adenosine monophosphate-activated protein kinase (AMPK) pathways, but can also mediate its signaling through the MAPK and PI3K-Akt pathways. Leptin action is inhibited by cytokine signaling 3 receptors (SOCS3). Through its action on PI3K-Akt and MAPK, leptin influences vascular tone and promotes endothelial and smooth vascular muscle proliferation and migration. Chronic hyperleptinemia also increases ROS production and decreases NO levels in endothelial cells, leading to vascular endothelial dysfunction. This effect is compounded by leptin-induced upregulation of endothelin-1, a potent vasoconstrictor, along with multiple mediators of vascular inflammation such as TNF-α, monocyte chemotactic protein-1 (MCP-1), IL-2, and IL-6 from endothelial cells and peripheral blood mononuclear cells, increasing risk of atherogenesis.
Adiponectin elicits anti-inflammatory and anti-atherogenic properties by multiple mechanisms of action that are lost in obesity/IR and may contribute to the development of diabetes and its cardiovascular complications [50, 51]. Perivascular adipose tissue senses β-adrenergic signaling that activates eNOS and NO production, which through its actions on mitochondria triggers adiponectin release into the circulation. Adiponectin binds to collagen, allowing it to accumulate in the subendothelial space of vascular walls where it represses the expression of adhesion molecules such as the intracellular adhesion molecule-1 (ICAM-1), vascular cell AM-1 (VCAM-1), and endothelial-selectin by inhibiting TNF-α and NF-kB. This minimizes monocyte attachment to endothelial cells and polarizes resident macrophages toward the anti-inflammatory M2 phenotype, thereby reducing foam cell formation by reducing cholesterol ester accumulation. Adiponectin also promotes endothelium-dependent vasodilation by binding adiponectin receptor 1 present in smooth muscle and endothelial cells, where it triggers NO formation and release through AMPK activation. NO in smooth muscle activates cyclic guanosine monophosphate (cGMP)-dependent protein kinase events that reduce intracellular Ca2+ levels (e.g., by opening of large calcium-sensitive potassium channels), leading to smooth muscle relaxation and vasodilation. Obesity/IR inhibits these vasculoprotective processes by blocking adiponectin signaling, impairing β-adrenergic receptor sensitivity, and reducing adiponectin and AMPK expression. As a consequence, blood vessels are vasoconstricted and more atherogenic, which compounded by hyperglycemia and dyslipidemia potentially increases cardiovascular risk.
Dyslipidemia and Vascular Disease in Diabetes
While hyperglycemia and adipokine imbalance clearly contribute to the pathogenesis of vascular disease in diabetics, lipid abnormalities such as decreased serum HDL-C, elevated sdLDL-C, and hypertriglyceridemia are believed to be of primary importance in precipitating coronary artery disease in diabetics [47]. Visceral adiposity, IR, and enhanced SNS activity associated with the MetS result in a chronic elevation of serum free fatty acids, which enhances hepatic VLDL-C and triglyceride (TG) synthesis and secretion. Prolonged elevation of VLDL and TGs, particularly postprandially due to IR, promotes sdLDL-C production in diabetics by enriching LDL particles and TGs that are subsequently modified by hepatic lipase and cholesterol ester transfer protein [52]. sdLDL-C is more susceptible to oxidation and adhesion to the vascular endothelium than LDL-C, making it particularly atherogenic and highly predictive of vascular endothelial dysfunction in diabetics [53]. Elevated TGs and hepatic lipase activity also modify the size, composition, and catabolic rate of HDL-C, ultimately reducing levels of this anti-atherogenic lipoprotein. Obesity/IR alters several other enzymes and regulatory pathways involved in hepatic and intestinal lipoprotein synthesis and release that are beyond the scope of this chapter (see [54] for review), which combine to make the mechanisms of dyslipidemia in diabetes particularly complex. Figure 3.2 presents an overview of the aforementioned mechanisms.
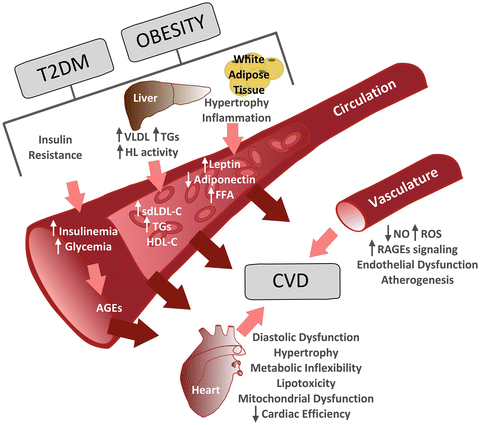
Fig. 3.2
Overview of the mechanisms and players involved in type 2 diabetes-related cardiovascular disorders. T2DM type 2 diabetes mellitus, VLDL very low-density lipoprotein, TGs triglycerides, FFA free fatty acids, sdLDL-C small-dense low-density lipoprotein cholesterol, HDL-C high-density lipoprotein cholesterol, AGEs advanced glycation end products, NO nitric oxide, ROS reactive oxygen species, RAGEs advanced glycation end products receptor, CVD cardiovascular disorders
Diabetic Cardiomyopathy
IR and MetS are independent risk factors for heart failure, and diabetes patients are up to five times more likely to have heart failure than the general population. In addition to the well-established pathogenic effects of hyperglycemia and hyperlipidemia on the coronary vasculature, diabetes and MetS are associated with a distinct form of cardiac dysfunction known as “diabetic cardiomyopathy” (DMCM) that occurs independent of the precipitating effects of coronary artery disease, hyperlipidemia, or arterial hypertension [55]. The etiology of this condition involves the complex interaction of metabolic, neurohormonal, and subcellular derangements that directly affect myocardial cells prior to the onset of overt diabetes or cardiac dysfunction. This section of the chapter will summarize the accumulating evidence from basic, translational, and clinical studies for the distinct effects of diabetes and MetS on the heart that support to the increasing consensus that novel therapeutic approaches may be needed to prevent and optimally manage heart failure in this patient population.
Organ Level Changes
The early stage pathology of DMCM is an increase in left ventricular (LV) wall thickness associated with interstitial accumulation of AGEs and reduced compliance [56], which later progresses to involve interstitial and perivascular fibrosis and cardiomyocyte hypertrophy. These changes contribute to an impairment of LV diastolic function, which is typically the first detectable sign of cardiac dysfunction in diabetes and MetS. Hyperglycemia is postulated to play an important role in this early fibrosis by increasing AGE formation, which promotes myocardial collagen deposition and pro-inflammatory signaling. This may be compounded by impaired myocardial energetics, lipid droplet formation (steatosis), and cardiomyocyte damage or loss due to oxidative stress and apoptosis (discussed below). Diastolic dysfunction may progress toward LV dilatation and compromised systolic function as is seen in classic heart failure pathogenesis, perhaps accelerated by alterations in myocardial metabolic substrate utilization, mitochondrial dysfunction, and reduced cardiac efficiency.
Toxic Effects of Hyperglycemia and Hyperinsulinemia
Hyperglycemia is a hallmark of diabetes and has been associated with elevated heart failure risk in both diabetic and nondiabetic populations [57]. It is widely considered to be a central initiating factor in the development of DMCM by increasing cardiac oxidative stress, inflammation, and maladaptive remodeling by activating pathogenic signaling by the myocardial renin-angiotensin system, transforming growth factor (TGF)-β/insulin-like growth factor 1 (IGF-1) (via hyperinsulinemia), and poly(ADP ribose) polymerase (PARP). A primary mediator of glucose toxicity in the heart is likely the formation of AGEs, which accumulate in tissues of diabetic patients and are thought to play an important role in diabetic complications. The precise chemistry and cellular actions of AGEs are incompletely understood, but it is generally accepted that their formation begins by nonenzymatic binding of glucose to amino acids or deoxyribonucleic acid (DNA), ultimately generating a variety of compounds that act through RAGEs to promote production of ROS, crosslink collagen, and induce pro-inflammatory (e.g., NF-κB) and hypertrophic (e.g., extracellular signal-regulated kinases [ERK]/MAPK) signaling [49].
A common pathogenic link among these systems activated by hyperglycemia is thought to be the generation of ROS by a variety of sources including NADPH oxidase, uncoupled eNOS, and mitochondria, perhaps compounded reduced cardiac antioxidant enzyme capacity. The ensuing oxidative and nitrosative stress can damage cellular proteins and lipids, leading to impairments in Ca2+ homeostasis, myocardial impulse conduction, and microvascular function [58]. Adding further insult to injury, hyperglycemia, in part by inducing hyperinsulinemia, also impairs cardiac autophagy by inhibition of AMPK and activation of mammalian target of rapamycin (mTOR) signaling, resulting in an aggregation of damaged proteins and cellular components in the diabetic heart [59]. This loss of cellular “quality control” has been linked to endoplasmic reticulum (ER) stress and mitochondrial dysfunction, further contributing to myocardial dysfunction and disease progression.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


