Figure 3–1.
Concomitant with the formation of chambers (atria, ventricles), an adult type of electrocardiogram (ECG) can already be monitored in the embryo [2]. Scanning electron microscopic photographs of the developing chicken heart with matching electrocardiograms. At H/H 18, locally fast-conducting chamber myocardium has differentiated as reflected in the electrocardiogram. A atrium, avc atrioventricular canal, oft outflow tract, V ventricle. Note that the ECGs are displayed mirrored to match with the position of the chambers in the embryonic heart. The apparent T-wave has not been labeled since it reflects the depolarization of the muscularized outflow tract at this stage rather than the repolarization of the ventricle
Early Peristaltic Hearts
A typical circulatory system is made of pumps and transporting vessels. Nature uses two different schemes to make muscle-pumping devices. In one version, also utilized by the intestine, peristalsis is the driving force. In contrast, in adult vertebrates an alternative version involving chambers and valves is used (see next section). In the peristaltic version, a wave of contractions runs along the muscle mantel enfolding the main blood vessel, and this action pushes the encompassed fluid ahead in either direction. Such a system is not particularly efficient, but it allows the steady movement of fluids and slurries. During evolution, polarity evolved in the primitive peristaltic chordate hearts and this resulted in dominant pacemaker activity at one end of the cardiac tube, transforming such a heart into a one-way pump. All regions of peristaltic hearts possess poorly coupled cells and intrinsic automaticity, by which depolarizing impulses propagate slowly along the tube, resulting in matching peristaltic waves of contraction [5–9]. The advantage is that hearts using these slow contractions do not require well-developed contractile structures like those present in the chamber myocardium of higher vertebrates.
Development of Chambered Hearts
It is important to appreciate that the basic characteristics of muscle cells comprising a peristaltic heart are similar to those comprising the nodes of a chambered heart [4], as this facilitates the understanding that the design of chambered hearts is derived from the peristaltic heart. Though they share design characteristics, the chambered heart and its associated functional requirements are obviously far more complex than a peristaltic heart. Chambered hearts are the more powerful hearts that can cope with the increasing demands imposed by a growing microcirculatory resistance due to the evolutionary development of liver and kidneys. To achieve this, the atria became the drainage pool of the body to allow efficient filling of the ventricles, while the ventricles themselves became the power pumps. Like peristaltic hearts, chambered hearts are directional because dominant pace making activity remains localized at the intake of the heart. A logical further addition to these chambered hearts was the development of one-way valves at both the inflow and the outflow of a chamber. This permitted that, with relaxation, a chamber could be prevented from refilling from the downstream compartment, and prevented, with contraction, backflow into the preceding compartment. Remarkably, the areas in which these valves evolve display many nodal characteristics, and are the same areas that in the vertebrate embryonic heart will not, or will only later develop into chamber myocardium [10]. Thus, cardiac valves are always found in regions of nodal-like myocardium. This holds true for the sinuatrial region, atrioventricular junctional region and also for the myocardial outflow region of the embryonic heart. Interestingly, the outflow tract myocardium in human can extend as far downstream as the semi-lunar valves. Spontaneous activity and even tachycardias originating from this area have been reported [11] underscoring the notion that this myocardium is persisting embryonic nodal-like tissue.
Development of the Cardiac Chambers and Conduction System
The four-chambered heart of mammals develops from a single tube, which initially contains the precursors for the left ventricle only, or even less. With development, the precursors for the remainder of the heart are added at the arterial and venous poles (Fig. 3.2) [12, 13]. The heart tube continues to elongate as a result of the recruitment of myocytes and bends ventrally and rightwards in a process called looping, which occurs around mouse embryonic day 8.5–9 (E8.5–9), comparable to 23 days in human development [14–16]. At this time, the heart tube has started to form a distinct ventricular chamber at its original ventral side, while the future atrial appendages differentiate slightly later at the dorsal and caudal end [17–19].
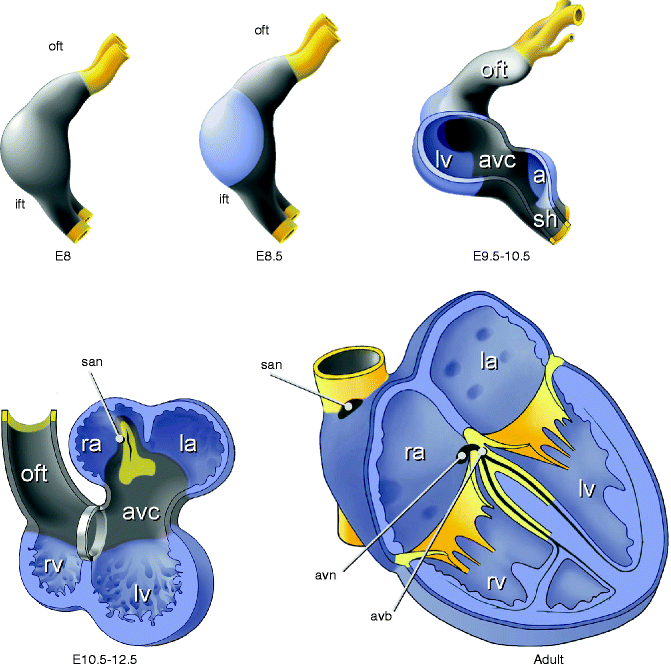
Figure 3–2.
Schematic overview of heart development in higher vertebrates. Chamber myocardium (blue) expands from the outer curvatures of the primary heart tube, whereas non-chamber myocardium (grey) of the inflow tract (ift), sinus horns (sh), atrioventricular canal (avc), outflow tract (oft), and inner curvatures does not expand. Sinus horn myocardium gives rise to the sinoatrial node (san), atrioventricular canal myocardium to the atrioventricular node (avn) and atrioventricular junction. First three panels show a left-lateral view of the heart
The myocardium of the original heart tube and the myocardium subsequently added to its poles, displays slow conduction and contraction, along with the property to spontaneously depolarize (automaticity), which is mainly Cx45 dependent [20], resulting in peristaltic-like, rhythmic contractions. Dominant pacemaker activity is always located in the newly formed myocardium at the venous pole [21, 22]. This generates a unidirectional blood flow from the venous to the arterial pole. While the ventricular and atrial chamber myocardium differentiates and rapidly proliferates at specific sites within the heart tube, the remainder of the myocardium maintains the primary phenotype. Once chambers have developed, this primary myocardium can be recognized as the sinus venosus, the atrioventricular canal (AVC) and the outflow tract (OFT). The sinus node develops from a small component of the sinus venosus [22, 23] and the AVN and AV junction myocardium develop from the AVC [24, 25]. Both nodal components still display phenotypic features of the primary myocardium they develop from, such as automaticity, slow conduction and poorly developed sarcomeres and sarcoplasmatic reticuli [26]. The maintenance of the primary phenotype of the myocardium is fundamental and regulated by the transcriptional repressor genes Tbx2 and Tbx3 [3, 27], which are selectively expressed in the primary myocardium and developing and mature conduction system [16]. A deficiency of Tbx3 in the myocardium results in expansion of the expression of working myocardial gens Cx40, Cx43, Nppa and Scn5a into the sinus node domain. In contrast, forced expression of Tbx3 leads to development of ectopic functional pacemaker tissue [27]. Both Tbx2 and Tbx3 are required to pattern and form the AVC in a redundant fashion [28]. Thus, Tbx2 and Tbx3 inhibit differentiation into working myocardium, which allows for the development of components of the cardiac conduction system. In conclusion, the primary myocardium lies at the basis of the pacemaker tissues of the cardiac conduction system.
In contrast, formation of the chambers is marked by the differentiation of localized regions of primary myocardium of the embryonic heart tube into the fast-conducting working myocardium of the chambers. A hallmark feature of this working myocardium is the expression of a chamber-specific gene program [4]. This includes genes for rapid propagation of the action potential such as Cx40 and Cx43 and the secreted factor Nppa (Natriuretic precursor peptide type A, also known as Anf). Moreover, working myocardium expresses the α-subunit of the sodium channel Nav1.5, encoded by SCN5a and develops a functional sarcoplasmatic reticulum [29]. At the cellular level, a local increase in cell size followed by local re-initiation of proliferation marks this area [30]. At the morphological level, initiation of chamber formation is marked by the appearance of trabecules, sponge-like myocardial structures developing at the luminal side of the outer curvatures of the developing heart, particularly in the future ventricular chambers. This series of events has been dubbed the ballooning model of chamber formation [4]. Both the chamber-specific gene program and gene expression in the maintained primary myocardium are regulated amongst others by cardiac transcription factors such as Nkx2-5, Gata4 and a number of T-box transcription factors (Tbx), such as Tbx5 and Tbx20 [15]. Disruption of any of these crucial factors leads to misspecification of working myocardium or primary myocardium which, in turn, eventually can result in local heterogeneities and arrhythmias.
T-box Transcription Factors Regulate Compartmentalization of the Heart
An important question is why some areas of the (embryonic) heart do not participate in the formation of atrial and ventricular working myocardium and mature in a nodal direction, such as the sinus venosus and the atrioventricular canal. To gain insight into this process we studied the regulation of the Nppa gene in more detail. Nppa is never expressed in nodal tissues from fish to human, and in the embryonic heart it marks the developing atrial and ventricular working myocardium [31]. While investigating the mechanism behind the chamber-specific expression of Nppa, we established that both a single TBE site (DNA binding/recognition site for T-box transcription factors) and adjacent NKE site (Nkx2-5 binding element), are present in the Nppa promoter and are required for repression of Nppa in the atrioventricular canal [3] and outflow tract [17]. T-box factors are evolutionary highly conserved transcription factors, which are important regulators of (cardiac) development. Presently at least 17 different T-box genes with diverse functions in development and disease are known [32]. In a search for the T-box factors that could act as a repressor for the Nppa gene, we observed that Tbx2 is expressed in inflow, atrioventricular canal, inner curvature and outflow myocardium. Moreover, expression of Tbx2 and Tbx3, a transcriptional repressor with a similar role, is confined to primary (non-chamber) myocardium, remarkably mutually exclusive to Nppa, Cx40, Cx43, and other chamber-specific genes [3, 33, 34]. These findings point to a model in which chamber formation (e.g. atria, left and right ventricle) and differentiation is driven by broadly expressed factors, in addition to which a supplementary layer of localized repressors inhibits this process in regions where chambers do not develop [35]. Tbx2 gain and loss of function experiments have demonstrated that Tbx2 is indeed able and required to inhibit chamber formation and expression of chamber marker genes [3, 36]. Tbx3 is expressed in a sub-domain of the Tbx2 domain, and whereas it is able to block chamber formation when expressed ectopically, its deficiency does not lead to obvious defects in atrioventricular canal patterning, indicating functional redundancy with Tbx2.
But how do Tbx2 and Tbx3 exert their functions? Both factors act as repressors of transcription and share DNA binding properties and target genes [37–41]. They effectively compete with Tbx5, a transcriptional activator, for TBE-binding, and for Nkx2–5 on NKE binding, thereby repressing chamber-specific genes and chamber differentiation [3, 34, 35]. Interestingly, lineage studies indicate that the sinus node develops from Tbx18-expressing precursors at the junction between the right sinus horn and the right atrium, whereas the atrioventricular node develops from the atrioventricular canal [23, 28]. The sinus node precursors induce expression of Tbx3, whereas the atrioventricular node precursors express both Tbx2 and Tbx3. Tbx2 is absent from the sinus node and sinus venosus and during development, Tbx2 becomes down-regulated. Tbx3 expression is maintained specifically in the nodes, thereby providing the only transcription factor found to date to be expressed specifically in the nodes (Fig. 3.3) [34, 36]. As mature nodes display many features that resemble primary myocardium in the embryo, it is attractive to hypothesize that their formation is the result of repression by Tbx2 and Tbx3 maintaining the primary phenotype.
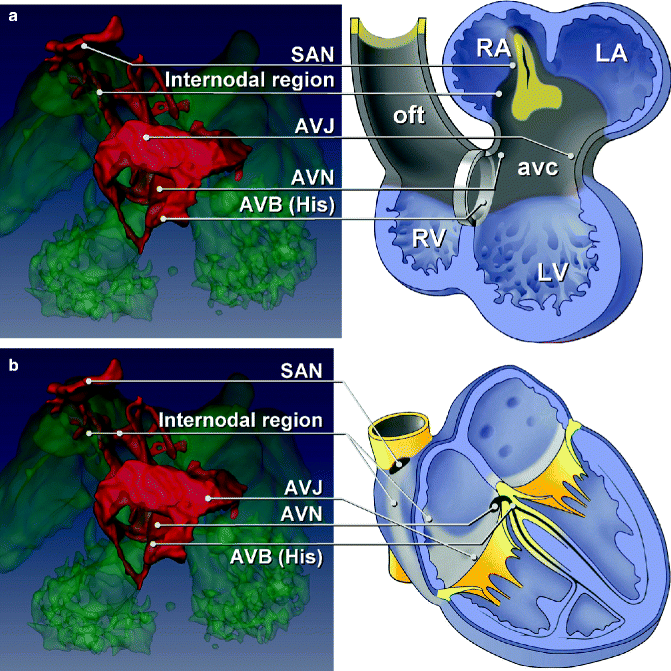
Figure 3–3.
Tbx3 (red) expression visualized in the heart at ED10.5 (a). Tbx3 marks the sinoatrial node, internodal region, AV junction, AV node and the AV bundles with respect to the location of the atria, the atrioventricular canal, the ventricles and the outflow tract (b) and with respect to the adult heart
Concluding, in a generalizing view one may envision that Tbx2, Tbx3 and/or other transcriptional repressors suppress the chamber-specific program of gene expression, allowing the regions where these factors are expressed to further mature into the nodal direction. Aberrant expression of such factors might thus be at the basis of ectopic automaticity and congenital malformations of the cardiac conduction system in the formed human heart.
Mutations in Transcription Factors Can Cause Congenital Heart Defects and Arrhythmias
As the above mentioned transcription factors are essential for the proper development of the cardiac (conduction) system, it is hardly surprising that mutations in these key genes lead to congenital heart defects. Mutations in Tbx5 in humans lead to the Holt-Oram syndrome (HOS), which is characterized by anterior pre-axial limb and cardiac malformations [42] (see below). Mutations in Tbx3 cause ulnar-mammary syndrome characterized by defects in breast development, apocrine gland, limb and genital formation [43], one study reporting ventricular septal defects and pulmonary stenosis [44]. Moreover, mutations in Tbx5 interacting partners such as Nkx2–5 and Gata4, like Tbx5 itself, are known to cause septum defects [45] and, in the case of Nkx2–5, atrioventricular conduction defects [46] (see below). Presently no diseases are known to be caused by Tbx2 mutations. A more common congenital disorder called DiGeorge syndrome, is caused by a 1.5–3 megabase genomic deletions of the chromosome 22q11 region which includes the Tbx1 gene [47]. Patients with DiGeorge syndrome are characterized by a variety of abnormalities including absence/hypoplasia of the thymus, cleft palate, facial dysmorphism and cardiovascular anomalies such as aortic arch malformation, outflow tract defects and VSDs. Recently it was postulated that a synergistic interaction between Tbx1 and Nkx2–5 might be responsible for the varying heart malformations of DiGeorge syndrome [48]. As, Tbx1-deficient mice phenocopy important aspects of DiGeorge including outflow tract abnormalities [49], it is believed that Tbx1 might modulate, in part, the outflow tract defects seen in DiGeorge patients.
Nkx2-5, AV Conduction and Atrial Fibrillation
One of the most intensely studied transcription factors involved in cardiac development is Nkx2-5. This homeobox transcription factor is expressed in the heart and other tissues and is part of the core cardiac transcriptional network essential for cardiac development. Targeted disruption of Nkx2–5 in mice causes early embryonic lethality, with cardiac development arrested at the linear heart tube stage, prior to looping [50]. Cardiac expression of Nkx2-5 continues throughout development and into adult life. In humans dominant mutations in NKX2–5 cause a variety of cardiac anomalies as well as atrioventricular abnormalities. Atrioventricular conduction abnormalities and atrial septal defects (ASD) are, in fact, the most common clinical presentations. However, other abnormalities such as ventricular septal defects, double-outlet right ventricle, tetralogy of Fallot and tricuspid valve abnormalities have also been noted [46, 51]. Cardiac dysfunction and sudden death have also been reported in mutation carriers. Atrioventricular conduction disease can occur in the absence of associated congenital heart defects [46, 51]. It is progressive in nature and electrophysiological studies have indicated that the conduction abnormality affects specifically the AVN. The fact that conduction defects can occur in the absence of cardiac structural malformations suggests that NKX2-5 has a function in conduction system development that is independent of its role in cardiac morphogenesis, and the fact that conduction disease is progressive with increasing age underscores the importance of normal NKX2–5 function in maintenance of AV conduction in adult life. Recent findings that genetic variation in the region of the NKX2.5 gene modulates the PR-interval in the general population seem to support such a role. The association signal detected at this locus however is located at some distance from the NKX2–5 gene and further research will be required to establish an unequivocal link between the genetic variation underlying this signal, NKX2-5 expression levels and the link to the PR-interval [52]. This is further underlined by studies using transgenic mice that carry a loss of function allele (DNA-non binding mutant) for NKX2–5. These transgenic mice were born with structurally normal hearts but displayed progressive atrioventricular conduction defects and heart failure. PR-prolongation was observed at 2 weeks of age, rapidly progressing into complete AV block by 4 weeks of age. A dramatic decrease in expression of gap junctional channels, Cx50 and 43, probably contributed to the conduction phenotype [53]. Geno-phenotype correlations on the various Nkx2-5 mutants in vitro suggested that the principle determinant of the two most common phenotypes (AV block, atrial septal defect) was the total dose of Nkx2-5 capable of binding to DNA [54]. It turned out that Nkx2–5 haploinsufficiency in the conduction system results in a significantly reduced number of normal cells, and that the specific functional defects observed in the knockout mice could be attributed to hypoplastic development of the conduction system. Thus, the postnatal conduction defects arising from Nkx2–5 mutations may results from a defect in the development of the embryonic conduction system. This conclusion was in line with experiments on heterozygous Nkx2–5 knockout mice with eGFP expression, knocked in the Cx40 locus, permitting the visualization of the conduction system. Indeed hypoplasia and disorganization of the Purkinje fibers was observed in the heterozygous Nkx2–5 mice and this was associated with abnormal ventricular electrical activation [55]. Analysis showed that maximal Nkx2-5 levels are required cell-autonomously and that a reduction in Nkx2-5 levels is associated with a delay in cell cycle withdrawal from surrounding Cx40 negative myocytes. This suggests that the formation of the peripheral conduction system is time- and dose-dependent on the transcription factor Nkx2-5, which is cell-autonomously required for the postnatal differentiation of Purkinje fibres. Experiments on Id2, a member of the Id gene family of transcriptional repressors, showing AVB-specific expression, proved that a critical transcriptional network, including Tbx5, Nkx2-5, and Id2, is required for the differentiation of ventricular myocytes into specialized cells of the conduction system [56]. Taken together, Nkx2-5 is required for development and homeostasis of atrio-ventricular conduction system (AVCS) and Purkinje fibers. In the case of the AVB some details of the molecular mechanism have been elucidated (Id2, Tbx5). It is noteworthy that Tbx3 also cooperates with Nkx2-5 [27] providing a link between this factor and Tbx3 in the AVB and AVN.
Some mutations in Nkx2–5 are not linked to AV block, but to atrial fibrillation [57], an arrhythmia long thought to have a possible origin in development [58]. In the majority of cases of paroxysmal atrial fibrillation, the myocardial sleeves surrounding the pulmonary veins at the orifice to the left atrium carry the triggers and possibly the substrate for the arrhythmia. Cells with presumed pacemaker activity have been found in the pulmonary veins of rat hearts [59] and in the pulmonary veins of patients with atrial fibrillation [60]. However, these observations are controversial, since lineage studies have demonstrated that the pulmonary myocardium is formed from a distinct lineage of precursor cells, different from the lineage that forms the muscle encompassing the sinus venosus (myocardium around caval veins, including the sinus node and around coronary sinus) [23, 61–63]. In contrast to the sinus muscle, pulmonary myocardium expresses from the outset transcription factor Nkx2-5 and its target gap-junction gene Cx40, suggesting it has an atrial working phenotype from the outset. Intriguingly, when Nkx2-5 protein levels are lowered experimentally, the pulmonary myocardium switches to a Cx40-negative, Hcn4-positive phenotype, similar to sinus nodal–like cells, resembling the sinus muscle. Thus, a reduction in the expression level of a single transcription factor, Nkx2-5, activates a gene program potentially sufficient to provide automaticity in the pulmonary myocardium. This observation suggests that inter-individual variation in Nkx2-5 dosage could be an important contributing trigger to the development of atrial fibrillation. Also, intriguingly, as mentioned above, genetic variation in the region of NKX2.5 has been associated with the PR-interval in the general population, which when prolonged, is in turn associated with risk of AF [64]. Taken together, variations in the (regulatory) sequence of the Nkx2–5 gene (and/or its interacting partners) are prime candidates for further research into the AV conduction and atrial fibrillation.
Tbx5, Atrial Fibrillation and Left-Right Chamber Differences
Another crucial factor in cardiac development is TBX5, which belongs to the evolutionarily conserved T-box family of transcription factors that bind DNA through a highly conserved DNA binding domain called the T-box. The consensus DNA sequences of the target genes where T-box factors bind are called T-box binding elements (TBEs) [65]. Tbx5 expression displays a posterior-anterior gradient along the heart tube with the most intense expression at the inflow tract of the heart. After chamber development has been initiated, Tbx5 expression is found in the sinus venosus, atria, AVC, left ventricle, including the left aspect of the ventricular septum, and to a lesser extent in the right ventricular trabecules [31, 66]. In humans, TBX5 haploinsufficiency has been shown to cause Holt-Oram syndrome (HOS), which includes congenital heart defects, conduction system abnormalities and upper limb deformities [42, 67]. Heterozygous Tbx5 knockout mice recapitulate many of the phenotypic abnormalities observed in Holt-Oram syndrome patients [68]. Expression of both Nppa and Cx40, targets of Tbx5, was also reduced in these mice during development. Complete deletion of Tbx5 in mice leads to embryonic death (around E9.5) with failure of heart tube looping and an under-developed caudal part [68]. Furthermore, Tbx5 overexpression in an embryonic cell line results in significant upregulation of Nppa and Cx40, and, counter intuitively, in cells displaying a spontaneous beating phenotype [69, 70]. Moreover, as mentioned earlier, Tbx5 is required for the differentiation of the AVB in the crest of the ventricular septum [71].
Recent work further links TBX5 to arrhythmogenic mechanisms and atrial fibrillation in particular. Atrial fibrillation has occasionally been described in sporadic patients with Holt-Oram [72], though principally in the setting of congenital heart disease and the resultant hemodynamic effects (atrial enlargement). We recently reported on a family in which affected patients have mild skeletal deformations (a hallmark feature of HOS) and very few have congenital heart disease. However, the great majority presented only with paroxysmal atrial fibrillation at an unusually young age [73]. Sequencing of TBX5 revealed a novel mutation, p.G125R, co-segregating with the disease. The mutant protein displayed significantly enhanced DNA-binding properties, which resulted in augmented expression of Nppa, Cx40, Kcnj2 and Tbx3 genes in comparison to wild-type TBX5, i.e. functioned as a gain-of-function factor. This is in contrast to all known Holt-Oram mutations to date, which result in a loss-of-function phenotype. This TBX5 gain-of-function mechanism probably underlies the paroxysmal atrial fibrillation and mild HOS phenotype. The mechanism underlying atrial fibrillation in TBX5 gain-of-function mutants may involve direct stimulation of TBX5 target genes involved in familial atrial fibrillation (NPPA, KCNJ2, CX40). Alternatively, it may involve TBX5-stimulated TBX3, as it was recently shown that TBX3 is highly sensitive to TBX5 dosage [74], and TBX3 controls the sinus node gene program and induces pacemaker activity in atrial muscle [27]. There is thus evidence in support of a role of TBX5 in the development of (paroxysmal) atrial fibrillation, a proposition that was recently further substantiated by the fact that genetic variation within the TBX5 gene was found to be associated with AF [75].
The left and right ventricle have a different morphology, tissue architecture, geometry, and function, and myocytes of the right and left ventricle differentiate to similar, but not identical phenotypes [76]. Differences in the regulatory and gene programs of progenitors of the left and right ventricle probably underlie the differences in these phenotypes. Tbx5 likely contributes to this, as it is expressed in an antero-posterior gradient in the heart tube regulated by retinoic acid [77]. The left and right ventricles are specified along the antero-posterior axis, and, as a consequence, the developing left ventricle robustly expresses Tbx5, in contrast to the right ventricle [66]. This suggests that Tbx5 is necessary for left ventricular identity, and provides, in part, the boundary between the left and right ventricle [78]. Consequently, target genes of Tbx5 are likely differentially regulated between the left and right ventricles. In fact, target gene Cx40, mimics the pattern of Tbx5 [68] and is differentially expressed between the left and right ventricle in the developing and adult heart. Seeing that the right ventricle especially is prone to the development of various arrhythmias, such as those seen in Brugada Syndrome [79] and Arrhythmogenic Right Ventricular Dysplasia (ARVD) [80], and given the fact that Tbx5 is actively involved in segregating between the left and right ventricle [78], it is tempting to speculate that target genes of Tbx5 might contribute to or oppose arrhythmias. Recent work has shown that numerous genes are influenced by differences in Tbx5 dosage, including genes expressed during heart development such as transcription factors (Tbx3, Irx2), cell-cell signalling molecules, and ion channels (Cx40, KCNA5) [74]. This, together with the link between TBX5 and atrial fibrillation, warrants further investigation into TBX5 and its downstream genes in relation to arrhythmias and ion channel genes.
Tbx2, Accessory Conduction Pathways and Defective Patterning and Gene Regulation Within the Atrioventricular Canal Myocardium
Wolff–Parkinson–White (WPW) syndrome is an arrhythmogenic defect characterized by a normal conduction system and one or more accessory AV connections, bypassing the AV node and His bundle, which cause ventricular preexcitation and predispose to re-entrant supraventricular tachycardia [81]. These abnormal accessory pathways may conduct faster than the AV node and this leads to ventricular preexcitation, evidenced by a short PR interval and a slurred upstroke of the QRS complex on the ECG. The majority of WPW occurs isolated and sporadic, affecting 1–3 persons per 1,000 [81], most patients with WPW syndrome have structurally normal hearts, though in a small portion of WPW patients, accessory pathways occur in association with congenital heart defects, such as Ebstein anomaly [82]. Nonetheless, autosomal dominant WPW families have been described [83, 84]. PRKAG2 and LAMP2 gene mutations were found in familial left ventricular hypertrophy and preexcitation, while a PRKAG2 mutation was detected in a single family with isolated WPW [85, 86]. Patients with mutations in the PRKAG2 gene have a variable combination of glycogen storage cardiomyopathy, progressive conduction system disease including sinus bradycardia and atrioventricular block, ventricular preexcitation, arrhythmias, and sudden death [85]. Ventricular preexcitation is presumably caused by a disruption of the annulus fibrosus, which electrically insulates the atrial and ventricular muscle masses, distinct from the muscular-appearing bypass tracts observed in typical WPW syndrome [87]. However, in a cardiomyocyte-specific overexpression model of mutated PRKAG, accessory pathways only developed when the mutated PRKAG was overexpressed during development. When overexpression started in adulthood, glycogen storage disease and conduction system degeneration occurred but accessory pathways did not develop, suggesting that a developmental defect may underlie pre-excitation in PRKAG mutants [88]. In addition, various NKX2–5 gene mutations in patients with CHDs are also associated with WPW, and point to a role of this critical cardiac transcription factor gene in WPW pathogenesis [51, 56].
A milder form of arrhythmias involving accessory pathways is the atrioventricular reentrant tachycardia (AVRT). This is the most common type of supraventricular tachycardia in both the fetus and the newborn [89]. Although AVRT can be potentially life-threatening and are sometimes difficult to control with antiarrhythmic drug therapy, in general these tachycardias resolve spontaneously within the first months of life, and >60 % of patients require no antiarrhythmic drug therapy and remain free of symptoms after the age of 1 year [90]. This self-resolving character of most perinatal AVRTs suggests that the majority of the accessory pathways eventually disappear after birth. Studies in animal models have shown that accessory AV myocardial connections are present until the late stages of cardiac development [91]. Moreover, a recent study on human hearts from neonates without episodes of supraventricular tachycardia found that isolation of the AV junction is a gradual and ongoing process [92]. The right lateral accessory myocardial AV connections in particular are commonly found at later stages of normal human cardiac development. These transitory accessory connections may therefore act as a substrate for AV reentrant tachycardias in fetuses or neonates.
The molecular mechanisms by which these accessory bypasses are formed are, however, poorly understood. The atrioventricular (AV) node and AV bundle are the only muscular connections that cross the annulus fibrosus. However, during heart development, in the absence of an annulus fibrosus, the AV canal myocardium still maintains adequate AV delay, allowing for the synchronized alternating contraction of the atria and ventricles [93, 94]. Remnant strands of this AV myocardium can however be observed in normal hearts around and after birth (see above). They disappear when the annulus fibrosus is fully formed [92, 95]. These strands are likely to maintain the slow conduction properties of the AV myocardium. Moreover, slow-conducting AV canal–type myocardium remains present around the orifices of the mitral and tricuspid valve in the adult heart [25, 96]. Together, this suggests that AV canal myocardium plays a central role in the AV delay and that defects in AV canal myocardium could underlie formation of functional accessory pathways. The AV canal myocardium is specified early in cardiac development by the expression of bone morphogenetic protein 2 (Bmp2) in the AV canal myocardium progenitors in the early heart tube, where it stimulates the expression of Tbx2 [97], a transcription factor required for the development of the AV canal [25]. Indeed in a recent study by Aanhaanen et al. [98], it was shown that myocardium-specific inactivation of Tbx2 leads to the formation of fast-conducting accessory pathways, malformation of the annulus fibrosus, and ventricular preexcitation in mice. The AV accessory pathways ectopically express proteins required for fast conduction (Cx40, Cx43, and sodium channel, SCN5A). Several other genes and pathways have been implicated in AV canal development such as the Bmp and Notch pathways. Interestingly, microdeletions of BMP2 and JAGGED1, a Notch ligand, have been associated with ventricular preexcitation in human [99, 100]. Moreover, deletion of the Alk3 receptor (activated by Bmp2) in the AV canal myocardium also results in accessory pathways and AV nodal defects in mice [101]. The fact that Notch signalling is involved in maturation of the AV canal-derived myocardium was underscored by another recent paper in which it was shown that activation of Notch signalling in the developing myocardium of mice produces fully penetrant accessory pathways and ventricular preexcitation [102]. Conversely, inhibition of Notch signalling in the developing myocardium resulted in a hypoplastic AV node, with specific loss of slow-conducting cells expressing Cx30.2 and a loss of physiologic AV conduction delay. In conclusion, these results indicate that defective patterning and gene regulation within the AV canal myocardium, via disruption of the AV canal regulatory network, leads to malformation of the annulus fibrosus, formation of accessory AV connections, and ventricular preexcitation.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


