4 Developmental and Pediatric Lung Disease
The diagnostic approach to pediatric lung biopsy differs somewhat from that in the adult patient. Many of the usual questions that arise in adult pulmonary pathology are replaced by separate issues involving abnormal development, altered lung growth due to prematurity, genetic disease, and infections secondary to an immature immune system.1 The spectrum of diseases observed in the pediatric lung biopsy differs from its adult counterpart and it is important to approach these biopsies with knowledge of lung development and anatomy. In addition, communication with the clinician, the radiologist, and the surgeon is essential, because a tentative list of diagnostic considerations usually can be built on the basis of clinical, radiologic, and intraoperative findings. This chapter covers a spectrum of common and rare entities that the surgical pathologist may encounter when examining biopsied and resected specimens from pediatric patients.
Processing of Pediatric Lung Biopsy Specimens
General processing of lung biopsy specimens is covered in Chapter 2. Recommendations have been published for processing of pediatric biopsy specimens for diffuse lung disease.2 A point worthy of emphasis is that in both diffuse and localized disease of the pediatric lung, infection should always be considered, and a substantial portion (one third to one half) of the surgical lung biopsy specimen should be sent for cultures if the surgeon has not already sent culture samples directly from the operating room. Touch imprints of the biopsy cut surface can be made and rapidly stained with silver stains for fungi or acid-fast stains for mycobacteria, a technique that is particularly useful for processing of lung biopsy specimens from immunosuppressed or immunocompromised patients. A few small pieces should be retained in glutaraldehyde for electron microscopy, which may have utility in the diagnosis of genetic disorders of surfactant metabolism and viral infections. Additional tissue should be snap-frozen and retained for potential molecular diagnosis of genetic disorders or infectious processes. For patients with suspected autoimmune diseases or chronic hemorrhage syndromes, a piece of lung tissue should be frozen in cryomatrix for possible immunofluorescence study. The remaining lung tissue should be expanded with formalin using a tuberculin syringe or other fine needle by transpleural injection, fixed for approximately 10 minutes, and sectioned for histologic examination. Inflation of pediatric lung biopsy is essential to reproduce the in vivo lung architecture and enables microscopic assessment of alveolar growth and development. In addition to standard hematoxylin and eosin (H&E) stains, many cases of diffuse disease benefit from the addition of connective tissue stains, such as trichrome, Verhoeff-van Gieson, or Movat pentachrome stains, for further evaluation of vascular disease or small-airway scarring, both of which may be under-recognized with use of routine stains. Additional staining for microorganisms, iron, glycogen, and alveolar proteinosis should be performed when indicated.
Cysts and Masses
Many pediatric lung biopsies and resections are performed for localized abnormalities within the lungs, and these may be solid or cystic (Box 4-1). A number of clues can be obtained from the history and findings on diagnostic imaging and intraoperative inspection, and these can be helpful in making the correct diagnosis, even before slides have been reviewed. The location of the mass, the presence of cystic or solid areas, the vascular and bronchial supply, and the onset of symptoms can all be useful in narrowing the scope of the differential diagnosis.3
Bronchogenic Cysts
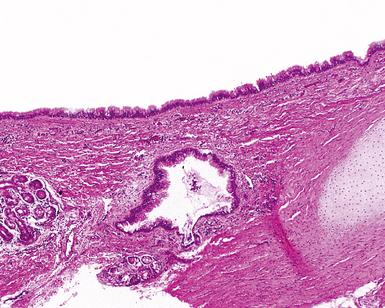
Bronchogenic cysts (or bronchial cysts) are developmental anomalies formed by abnormal budding of the tracheobronchial anlage of the primitive foregut in early development.4,5 They commonly are found in the anterior mediastinum or along the tracheobronchial tree. Less often, they are found within the pulmonary parenchyma, within or below the diaphragm, or even within the pericardium.6,7 Patients with bronchogenic cysts can present with infection or obstruction, although frequently these lesions are an incidental radiologic finding.5 The cyst often is unilocular and lined by ciliated columnar epithelium. Many bronchogenic cysts communicate with the tracheobronchial tree. On occasion, they show squamous metaplasia or mild chronic inflammation.
Considerations in the differential diagnosis for bronchogenic cyst include esophageal duplication cyst for lesions occurring in the mediastinum and congenital pulmonary airway malformations (CPAMs) for lesions occurring within the substance of the lung. The bronchogenic cyst typically has cartilage plates and submucosal glands in the wall, similar to the normal microscopic anatomy of bronchi (Fig. 4-1). These structures may be sparse but can help differentiate this lesion from the esophageal duplication cyst, which lacks these structures and has a double muscular layer in its wall. Both of these entities can be lined by ciliated mucosa. The bronchogenic cyst lacks connection with alveolar tissue, a feature that aids in its distinction from CPAM. On occasion, with inflamed cysts within the lung, the nature of the specific lesion or underlying disorder may be impossible to ascertain. In such situations, a generic diagnosis of “inflamed intraparenchymal cyst,” accompanied by a differential diagnosis summary that includes abscess, CPAM, bronchogenic cyst, esophageal cyst, and intralobar sequestration, is appropriate. Radiologic and clinical features may help in further differentiating among these entities.
Bronchial Atresia
Bronchial atresia is one of the most common forms of congenital pulmonary malformation. Atresia of a segmental or subsegmental bronchus classically results in a central mucus-filled cyst (mucocele) at the point of atresia, dilated distal airways with mucous plugs, and hyperinflated microcystic distal parenchyma (Fig. 4-2). Microscopically, the abnormally developed cystic parenchyma has a pattern identical to that described in type 2 CPAM, consisting of increased numbers of abnormal bronchiolar structures surrounded by variably abundant alveolar spaces which are typically distended and either round or elongated in shape.8,9 Abundant mucus and muciphages typically are noted within proximal airway lumens and adjacent air spaces. Bronchial atresia often is detected prenatally or in infancy as an asymptomatic cystic lesion. Because of interconnection with the normal parenchyma through alveolar pores of Kohn, the abnormal lung distal to a bronchial atresia may potentially become secondarily infected, and presentation in later childhood, adolescence, or adulthood typically is due to symptoms of recurrent pneumonia. The primary considerations in the differential diagnosis are congenital lobar overinflation, which is characterized by normally developed (albeit markedly distended) air spaces, and intralobar sequestration, which is distinguished by the additional finding of aberrant systemic arterial supply.3
Pulmonary Sequestration
Pulmonary sequestration refers to the occurrence of lung tissue that does not communicate with the tracheobronchial tree and that typically has a systemic, rather than pulmonary, arterial supply.10–13 Such lung tissue is therefore “sequestered” from the usual pulmonary airway and vascular connections. These lesions are further subdivided into an extralobar type, which occurs outside the visceral pleura of the adjacent lung, and an intralobar type, which resides within the visceral pleural investment of a lung lobe. Although there is general agreement that extralobar sequestrations represent congenital malformations, the origin of intralobar sequestrations has been more controversial. More frequent diagnosis in adults has led to the theory that they are post-inflammatory lesions with acquired loss of bronchial connection and development of collateral systemic circulation from hypertrophied pulmonary ligament arteries.11 However, intralobar sequestrations diagnosed prenatally or in association with congenital malformations support the concept that the pathogenic mechanisms for extralobar and intralobar sequestrations are similar, and that adult lesions may represent occult malformation detected later in life. Specific features of extralobar and intralobar sequestrations, summarized in Table 4-1, are discussed next.11
Table 4-1 Pulmonary Sequestration: Extralobar versus Intralobar
| Clinical/Historical Feature | Extralobar Sequestration | Intralobar Sequestration |
|---|---|---|
| Location | Outside pleura of lung (“accessory lobe”) Commonly, left lung base | Within pleura of lung lobe Lower lobe (98%) |
| Gross | Pyramidal structure | Congested, hemorrhagic segment(s) of lobe |
| Age at diagnosis | 60% < 6 months | 50% > 20 years |
| Arterial supply | Systemic | Systemic |
| Origin | Congenital anomaly | Congenital anomaly; possibly acquired in adults |
| Histologic appearance | CPAM type 2 pattern Hyperinflated air spaces | CPAM type 2 pattern with mucus stasis Hyperinflated, hemorrhagic segment(s) of lobe Inflamed, chronic pneumonia |
CPAM, congenital pulmonary airway malformation.
Modified from Stocker JT. Sequestrations of the lung. Semin Diagn Pathol. 1986;3(2):106–121; and Langston C. New concepts in the pathology of congenital lung malformations. Semin Pediatr Surg. 2003;12(1):17–37.
Extralobar Sequestration
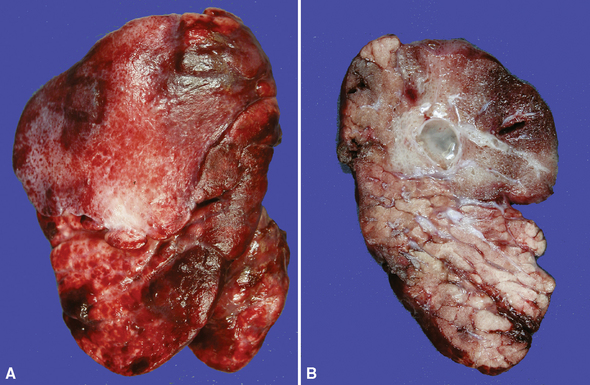
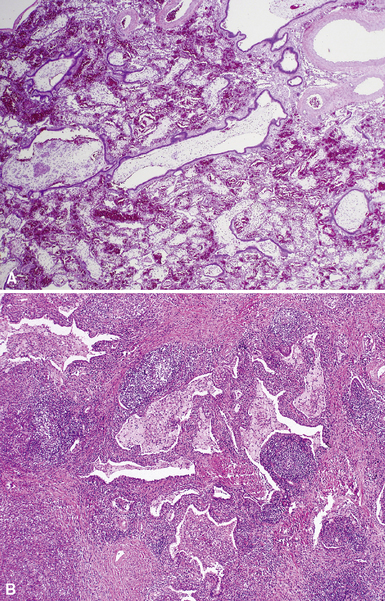
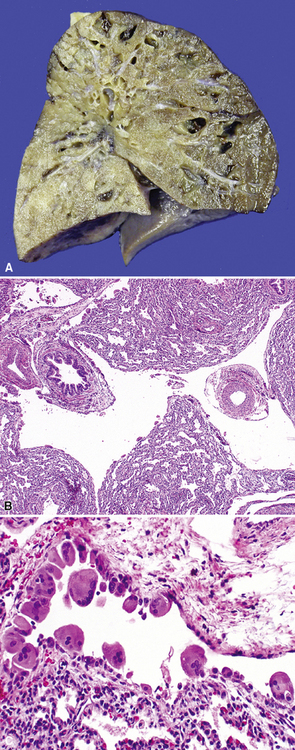
Extralobar sequestrations (ELSs) are thought to arise a a result of abnormal budding from the tracheobronchial anlage. Lying outside the normal lung, these structures appear as “accessory lobes,” completely surrounded by their own visceral pleura.10–15 ELSs usually are found in the lower thoracic cavity but may be found above, within, or below the diaphragm. On gross inspection, they appear as irregularly ovoid or pyramidal portions of lung tissue surrounded by pleura and with a vascular pole at one edge (see Fig. 4-3A). The radiographic or intraoperative finding of a systemic arterial supply confirms the diagnosis of ELS. The systemic artery can arise from a source above or below the diaphragm.16,17 The microscopic appearance may vary but the histologic pattern typically is that of type 2 CPAM and less often resembles that of normal lung11,14,15,18–21 (see Fig. 4-3B and C). Striated muscle occasionally is present within the interstitium of the lesion; this feature is called rhabdomyomatous dysplasia.
Intralobar Sequestration
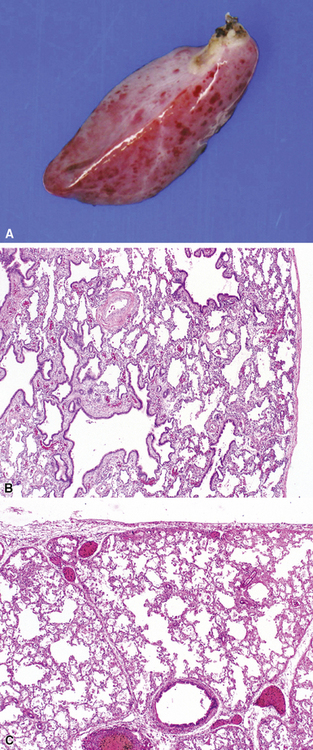
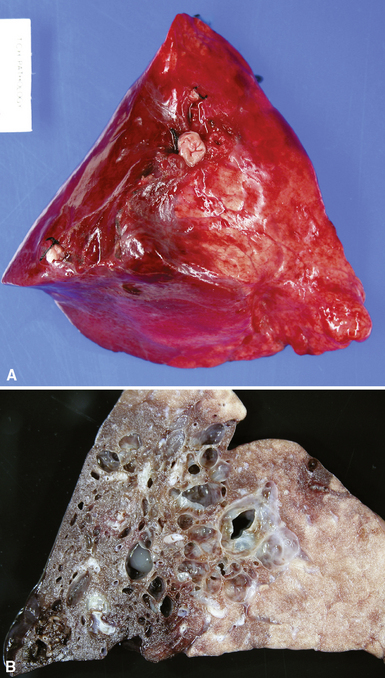
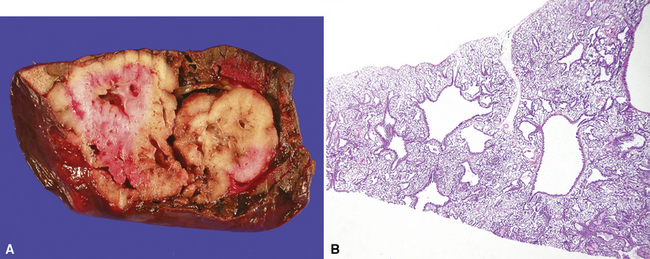
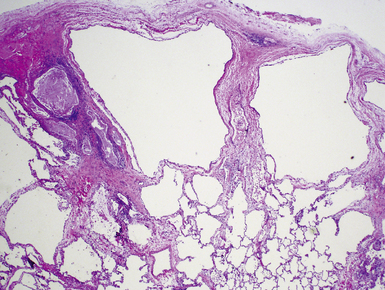
Intralobar sequestrations (ILSs) lie within the parenchyma of the lung. Most ILSs occur in the medial area of a lower lobe, and the abnormal systemic elastic artery can usually be identified in the region of the inferior pulmonary ligament22 (Fig. 4-4A). Radiologic studies can be extremely helpful in supporting the diagnosis.23,24 Various modalities, including computed tomography (CT) and magnetic resonance imaging (MRI), will show a solid or cystic mass that lacks normal bronchovascular patterns. A systemic arterial supply may be confirmed radiologically or at the time of surgery. The gross and microscopic findings are markedly influenced by age at resection and presence or absence of any accrued chronic inflammatory insults (usually secondary infections) within the sequestered lung tissue. In infants with asymptomatic lesions, the lobe contains a segmental region of congestion and hemorrhage due to the high flow of the systemic circulation, accompanied by microcystic parenchyma (see Fig. 4-4B). The maldeveloped parenchyma shows mucus stasis, identical to that in segmental bronchial atresia (Fig. 4-5A). In older children and adults with recurrent pneumonia, the histologic findings are similar in appearance to those in localized bronchiectasis with recurrent infection. Other features include marked acute and chronic inflammation with fibrosis and cyst formation (see Fig. 4-5B).
Congenital Pulmonary Airway Malformations
Congenital malformations of the pulmonary airways (i.e., CPAMs), also called congenital cystic adenomatoid malformations (CCAMs), are masses of maldeveloped lung tissue that are classified according to their gross and microscopic appearance.12,25–34 These lesions are identified most commonly in stillborn infants or in newborns with respiratory distress, but they can be discovered in adolescents and rarely in adults.35 Stocker and colleagues initially proposed a classification scheme for CCAM that divided these malformations into three subtypes.36 This scheme was later expanded to five subtypes (types 0 to 4), with a subsequent change in terminology from CCAM to CPAM, acknowledging that not all of these malformations are cystic and not all are adenomatoid.37 The overriding principle of this subclassification is that the dominant morphologic constituent of each type of CPAM reflects the morphology of the normal tracheobronchial tree from proximal to distal—that is, from malformed bronchi, to bronchioles, to distal lung (alveolar) tissue. This construct has provided useful morphologic distinctions and will continue to be revised as the pathogenesis of these lesions is better understood. CPAM classification may be difficult in immature or fetal lungs, and an alternate classification has been proposed for fetal lung resections.33,34
Type 0 CPAM, also called acinar dysplasia, is a rare lesion composed of cartilaginous airways and loose mesenchyme (see later discussion).38–41 Types 1 to 3 have a common general overall appearance of cystic spaces (distorted airways) with intervening structures more or less resembling alveoli (Figs. 4-6 to 4-8). Type 1 CPAM shows larger cysts, having some bronchial differentiation in that they contain ciliated epithelial lining or mucinous-type epithelium or have cartilage in their walls. Type 2 CPAM shows smaller cystic spaces that resemble ectatic irregular bronchioles, evenly separated from each other by alveolar structures, and represents a histologic pattern that implies intrauterine bronchial obstruction, typically from bronchial atresia or pulmonary sequestration. Type 3 CPAM is a rare solid lesion typically encompassing an entire lobe or lung and resembles pulmonary hyperplasia or immature lung in the early canalicular stage of development. Type 4 CPAM has been described as a peripheral large cyst with thin walls and flattened alveolar-type epithelial lining. Existence of this type is controversial, because it may represent unrecognized, undersampled, or completely differentiated examples of cystic pleuropulmonary blastoma (PPB).42–48 Large cysts resembling type 1 or type 4 CCAM should be extensively sampled to exclude the presence of small foci of either malignant primitive spindled cells beneath the alveolar epithelium (“cambium layer”) or immature chondroid foci, diagnostic of cystic (type I) PPB. Immunohistochemical staining for myogenin, desmin, and MyoD1 may be helpful in differentiating the cells of PPB from reactive fibroblastic proliferation or cellular mesenchyme in CPAM. Cystic PPBs have potential for recurrence as high-grade tumors, particularly if incompletely resected.
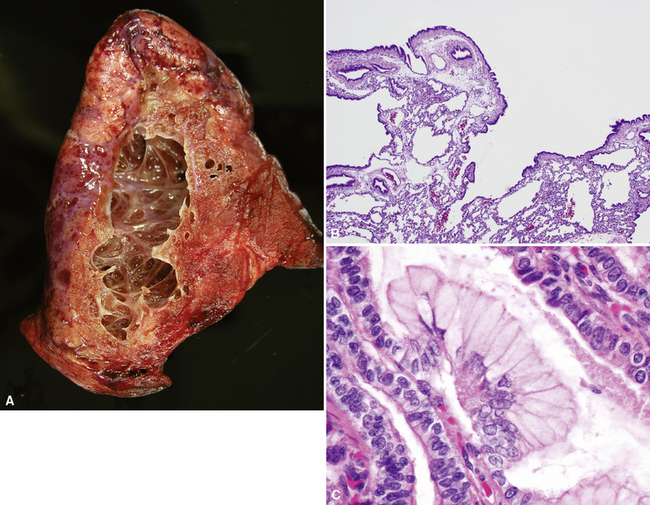
Although the prognosis in most cases is favorable following resection, there are rare reported cases of patients developing carcinomas in association with CPAM.49–55 Many of these lesions are mucinous bronchioloalveolar carcinomas, which has led to the proposal that the mucigenic epithelium in type 1 CPAM is preneoplastic (see Fig. 4-6C). Multifocal bilateral mucinous bronchioloalveolar carcinomas after incomplete resection of CPAM have been described in patients as young as 11 years of age.51 In light of these rare cases, the presence of mucinous epithelium in CPAM and completeness of resection should be documented for follow-up purposes.
Pulmonary Interstitial Emphysema
Pulmonary interstitial emphysema (PIE) results from dissection of air into the interstitial connective tissue of the lung. Rupture of alveoli or disruption of airway walls often is responsible for this phenomenon.12,56–59 Air accumulates in the interstitium along bronchovascular bundles and interlobular septa, creating cystic spaces that may at first resemble tissue architecturally torn during sectioning (Fig. 4-9A and B). The usual clinical scenario is that of a premature infant with neonatal respiratory distress syndrome receiving mechanical ventilation. Acute PIE usually resorbs over time, but the chronic form persists as cystic lesions lined by fibrous tissue or multinucleate giant cells (see Fig. 4-9C). Grossly, the process can involve both lungs diffusely or may be localized to one or two lobes. Multiple small cysts can be seen to extend along interlobular septa.
Peripheral Cysts Secondary to Lung Maldevelopment
Hypoplastic lungs, and those damaged in the neonatal period, are susceptible to persistent alterations in alveolar growth. This maldevelopment frequently appears as alveolar enlargement or cysts, particularly in the subpleural areas and peripheral lobules. Microscopic examination reveals irregular air space enlargement with fibrovascular walls lined by alveolar cells (Fig. 4-10). Peripheral cysts have been described in the lungs of several patients with Down syndrome.60–65
Pulmonary Hyperlucency
Several conditions may lead to the radiologic appearance of hyperlucency (Box 4-2). Clinical history and knowledge of the indication for resection are helpful, because the pathologic findings can be extremely subtle histologically. The clinical presentation may include shortness of breath, tachypnea, wheezing, or cough, typically in infants. The chest radiograph shows marked lobar enlargement with displacement of the mediastinum. The two most commonly occurring histologic patterns are congenital lobar overinflation (so-called congenital lobar emphysema) (in 70% of the cases) and polyalveolar lobe (30%).
Congenital Lobar Overinflation
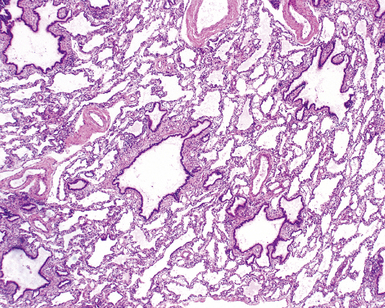
Congenital lobar overinflation (CLO), or congenital lobar emphysema, occurs when there is overdistention of the normal alveolar parenchyma12,66,67 (Fig. 4-11). The etiology is variable, but the underlying cause frequently is a partial or intermittent high-grade obstruction of the bronchus supplying the affected lobe.68 Bronchomalacia may result in collapse of the lobar bronchus with expiration, resulting in progressive air-trapping in the affected lobe. The obstruction can occur as a result of other intrinsic factors such as bronchial stenosis, abnormal “kinked” bronchial anatomy, mucosal webs, or mucous plugging. Alternatively, congenital lobar emphysema can be extrinsic as a result of various vascular or neoplastic etiologies. Approximately one half of the cases are idiopathic. The upper lobe is involved in nearly all cases. Lower lobe involvement is highly unusual except in acquired cases in patients with previous hyaline membrane disease or bronchopulmonary dysplasia (BPD). Some cases may arise secondary to trauma from tracheal suctioning during respiratory support.69
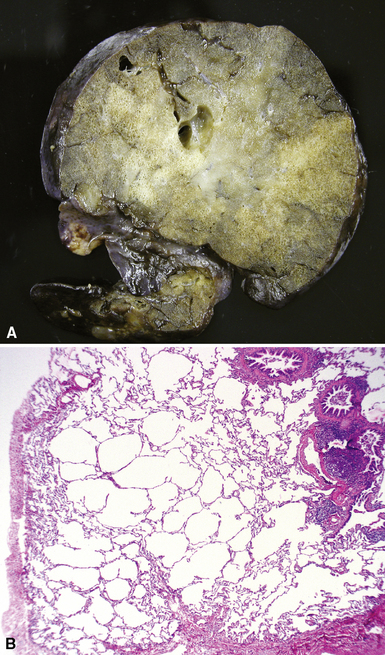
On gross examination, CLO is characterized by a markedly enlarged lobe, which generally retains its basic shape. Alveoli, alveolar ducts, and respiratory bronchioles typically are dilated on histologic examination. Unlike bronchial atresia and CPAM, CLO shows otherwise normal alveolar development, with appropriate numbers of bronchiolar structures and appropriate alveolar septation. The source of obstruction is identified only occasionally by gross and microscopic examination70–72 (Fig. 4-12).
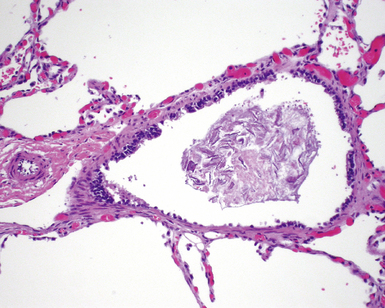
Polyalveolar Lobe
Polyalveolar lobe occurs when there is an increase in the regional number of alveoli relative to the corresponding conducting airways and arteries.73–75 Whereas the arteries and airways in these lungs are normal, the alveolar regions are enlarged by an increased number of nearly normal alveoli. The diagnosis can be made by radial alveolar counts, which are performed by counting the number of alveoli transected by a line drawn from the respiratory bronchiole to the nearest acinar edge (pleura or septum).76 The normal count varies with age but should be between 5 and 10 for infants, and 10 and 12 for young children. Radial alveolar counts in polyalveolar lobe will be approximately two to three times that number (Fig. 4-13).
Disorders of Lung Development
Acinar Dysplasia
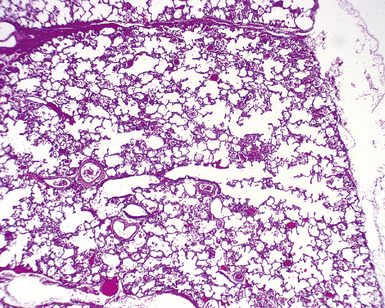
Described in 1986, acinar dysplasia is a rare severe diffuse developmental lung disorder resulting in marked deficiency of acinar development, identical to the entity described as type 0 CPAM.38–41 The lungs are small, with accentuation of small lobules by white interlobular septa (Fig. 4-14A). Microscopically, the lobular bronchi are surrounded by only few primitive air spaces, with virtually no alveolar development (see Fig. 4-14B). This disorder is uniformly fatal within the first few hours of life and typically is diagnosed at autopsy. Although acinar dysplasia is presumed to be of genetic origin, the etiology is unknown.
Congenital Alveolar Dysplasia
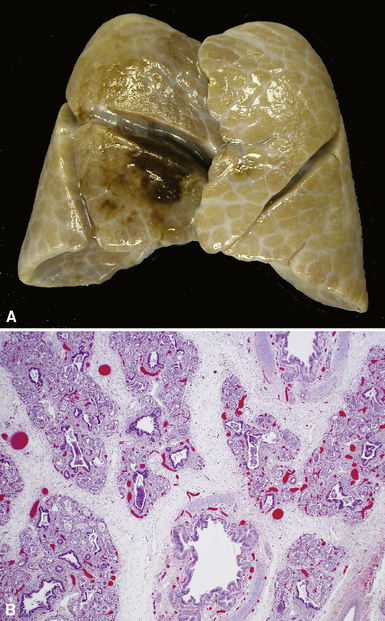
Congenital alveolar dysplasia also is a rare diffuse developmental lung disorder with incomplete air space development.77 Relative to those in acinar dysplasia, the air spaces are more numerous and exhibit greater complexity, but completely mature alveoli are lacking. The air spaces show primary septation but insufficient secondary septation, generally resembling the saccular stage of development (Fig. 4-15). This disorder can be difficult to distinguish from prematurity of lungs with superimposed injury and remodeling due to prolonged ventilation, and on a practical level, the diagnosis is reserved for term infants, in whom the disorder is more likely to be a developmental abnormality, rather than an acquired impairment of alveolar growth. The etiology is unknown.
Pulmonary Hypoplasia
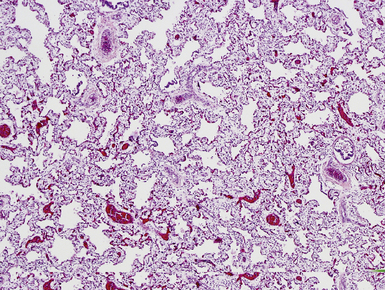
Pulmonary hypoplasia refers to abnormally small size of the lungs due to limitation of intrauterine development. Although primary forms of hypoplasia exist, this term most commonly refers to the secondary forms of lung hypoplasia in which physical compression of the lungs, by extrathoracic or intrathoracic processes, limits intrauterine growth. Common causes include congenital diaphragmatic hernia (Fig. 4-16
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


