Determinants of Cardiac Remodeling and Progression to Heart Failure
Jean-Jacques Mercadier
The pathways and mechanisms of cardiac hypertrophy and progression to heart failure (HF) have been for decades (if not centuries) questions challenging the intelligence of physiologists and physicians around the world (1,2,3,4,5). The question of the adaptive nature of cardiac hypertrophy arose logically when its etiologies (mainly hypertension and other causes of chronic mechanical overload of the heart such as valvular heart diseases) were progressively deciphered and when Laplace’s law was transposed to cardiac physiology and pathophysiology. According to Laplace’s law, the increase in wall thickness of the left ventricle (LV) allows normalization of LV wall stress during both systole and diastole, thus allowing each cardiac myocyte to operate under a normal load (i.e., normal working conditions). This concept allowed Meerson (1) to describe the time course of heart diseases due to LV chronic hemodynamic overload as three successive phases according to the degree of normalization of this load and what was considered at that time as the resulting LV function. During the first phase that immediately follows the imposition of the overload to the LV, the load by definition exceeds LV muscle mass, resulting in decreased LV output and the activation of a number of local and systemic processes recruited that emergently maintain mean blood pressure in the short term, and to normalize LV working conditions (i.e., ventricular wall stress) in the long term. If these mechanisms are successful, the patient (or animal in the experimental setting) survives, allowing the occurrence of the second phase of compensated/compensatory LV hypertrophy (LVH) during which the development of myocardial hypertrophy serves to normalize LV wall stress (i.e., has more or less exactly compensated the increase in load, allowing normalization of LV function and therefore, cardiac output) (6). The third phase returns full circle to the first one, during which LV load again progressively exceeds its contractile capacities allowed by a given myocardial mass and contractile performance that progressively declines. This results in decreased cardiac output and marks the entry to the chronic HF phase.
In this scheme, LVH is, by definition, adaptive. Decompensation occurs when the progression of load due to causative disease leads to deterioration of LV contractile function and to the increasing activation of the neurohumoral systems that surpass the growth capacity of the myocardium. This has been the dominating paradigm for almost four decades (Fig. 3-1).
However, since the beginning of the 1980s the adaptive nature of the second phase of compensated LVH has been challenged by two sets of concepts. The first arose from the demonstration that, beyond the increase in LV mass and an apparently conserved LV contractile function, the LV undergoes a profound tissular, cellular, and molecular remodeling. The latter is characterized by the expression of a protein phenotype typical of the embryonic/fetal stages of cardiac development [expression of β-MHC (Myosin Heavy Chain) instead of α-MHC, high levels of expression of natriuretic peptides, low level of expression of the Ca2+-ATPase of the sarcoplasmic reticulum (SERCA), etc.] known as the re-expression of the fetal gene program (FGP) (7). Although the α- to β-MHC transition could be considered, from an energetic perspective, as an adaptive process at the myocyte level, and the ventricular recruitment for the production of natriuretic peptides as an adaptation of the LV pump in face of an increased preload, the truly adaptive and beneficial nature of other changes such as decreased SERCA expression and pump function, cardiac myocyte apoptosis, or LV interstitial fibrosis were obviously questionable. The second set of concepts arose
from clinical research demonstrating that cardiac hypertrophy, even during its so-called compensated/compensatory phase, is a risk factor for HF and sudden death (8,9).
from clinical research demonstrating that cardiac hypertrophy, even during its so-called compensated/compensatory phase, is a risk factor for HF and sudden death (8,9).
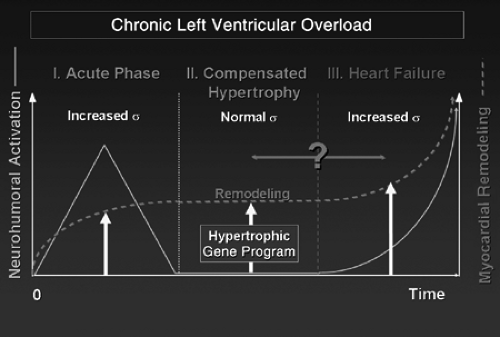 Figure 3-1 Progression of chronic work overload of the left ventricle from the onset of overload to chronic heart failure. This scheme is best-suited for myocardial infarction or experimental animal models in which the onset of the overload is sudden. During the acute initial phase, alteration of LV pump function results in activation of systemic neurohumoral systems aimed at maintaining mean blood pressure as normal as possible. Together with LV mechanical overload (mainly diastolic stretch), neurohumoral activation immediately triggers myocardial cellular and molecular remodeling characterized by the expression of the hypertrophic (fetal) gene program. Such a process persists during the second phase of compensated hypertrophy despite possible normalization of wall stress (sigma) and disappearance of systemic neurohumoral activation. This may be due, at least in part, to incomplete normalization of wall stress and to tissular neurohumoral activation. A strict limit between the second and third phases is hypothetical (?), progression to heart failure being progressive with gradual reappearance of decreased LV pump function. This decrease, associated with progressive increase in LV dimensions, reactivates mechanical overload and systemic neurohumoral systems, futher boosting myocardial remodeling. |
Finally, owing to the availability of more and more sophisticated transgenic (TG) mouse models and new tools to decipher the complexity of intracellular signaling, the last 10 years have been a time of a profound conceptual revolution regarding cardiac hypertrophy, its determinants, pathways, and biological significance. In particular, it has become progressively clearer that besides the adaptive nature of LVH, detrimental processes occurring from the very beginning of the overload and worsening during the phase of compensated/compensatory hypertrophy were, at least in part, responsible for the progression to HF. Thus, modification of the initial paradigm allows one to describe a continuum of myocardial, cellular, and molecular remodeling processes and pathways operating from the very beginning of LV reaction to biomechanical stress. It also sets the stage to distinguish between those processes governing physiological, beneficial, and/or adaptive responses from pathological, detrimental, and/or maladaptive hypertrophy. The purpose of this chapter is to review the determinants of this paradigm, with a special focus on the processes involved in the essentially pathological, detrimental, and/or maladaptive hypertrophy governing the progression to HF and the possible new therapeutic targets thus revealed.
Experimental Models and Initiators of Cardiac Hypertrophy
Experimental Models
It is classical to distinguish between two different LV hypertrophic phenotypes with distinct triggers: concentric hypertrophy due to pressure overload (PO) and eccentric hypertrophy due to volume overload. The latter is also seen after large myocardial infarction (MI). Accordingly, experimental models of hypertension/PO of the LV are used to reproduce concentric hypertrophy and models of valvular regurgitation, or high-output arteriovenous fistula are used to reproduce eccentric LV hypertrophy (10). A detailed description of the various models of hypertension has been produced recently by Pinto et al. (11), who elegantly compared the advantages and disadvantages of each model regarding end-organ damage, among which is LVH, eventually followed by HF. It should be emphasized that despite a number of common features (hypertension, LVH, and impaired endothelium-dependent vascular relaxation), each model also more or less specifically imprints LV remodeling, leading to more- or less-specific cardiac phenotypes. It must also be pointed out that the models used for hypertension research are not necessarily the same as those used for research on cardiac hypertrophy and the mechanisms of its development. In the latter context, most of the studies carried out during the 1980s and early 1990s have used (probably because of the simplicity of its performance) the model of constriction of the abdominal aorta (AAC). This utilizes a metal clip or a thread placed around the abdominal aorta, usually between the renal arteries (10), making it a model similar to the 2K1C Goldblatt model because of its early activation of the renin-angiotensin system (RAS) (12). But constriction of the thoracic aorta (TAC) has also been used, although with some differences with AAC regarding pattern of molecular remodeling (13). Around the mid-1990s transgenesis opened new avenues for research in the mechanisms of LVH and progression to HF. Since that time, each model has been developed to address a specific question related to the role of proteins involved either in myocyte function (such as phospholamban) (14) or to signaling pathways involved in the development of LVH (15). The extremely numerous transgenic mouse models cannot be detailed here but, rather, specific models will be referenced throughout this chapter, wherever necessary.
At this stage, it is important to distinguish between two different types of models: those spontaneously exhibiting a clear cardiac phenotype such as occurs with Gαq (15) or calcineurin (16) overexpression, and those with no cardiac phenotype, for which a mechanical stress (usually TAC-induced PO) is necessary to unmask a cardiac phenotype or to induce progression from compensated LVH to HF (17,18,19). Both types of models have markedly contributed to the elucidation of molecular mechanisms responsible for LVH and progression to HF. It is important to note that with the transition from rat to mouse, AAC has been progressively abandoned in favor of TAC. Except for when the clip is placed in young animals with little or no initial aortic constriction (but with constriction developing progressively with time as animals grow), the mechanical component of
the load imposed to the LV is much greater in the latter model than in the former, in which activation of the RAS seems to play a more important role (13,20). Therefore, it is clear that data obtained during the 1980s and 1990s using AAC in rats cannot be directly compared with data obtained more recently using TAC in mice.
the load imposed to the LV is much greater in the latter model than in the former, in which activation of the RAS seems to play a more important role (13,20). Therefore, it is clear that data obtained during the 1980s and 1990s using AAC in rats cannot be directly compared with data obtained more recently using TAC in mice.
Finally, the model of MI must be considered in the context of HF. Even if MI is an invaluable model to reproduce chronic HF (which is most often congestive in the setting of experimental models), allowing studies of peripheral adaptation or maladaptation or pharmacological studies, MI is not an appropriate model to identify the determinants of myocardial hypertrophy and the progression to HF. Indeed, LV remodeling following MI is more complex than that seen in models of pure pressure or volume overload, as the increase in systolic and diastolic stress on the noninfarcted myocardium depends on scar size and associated LV shape changes (21,22,23,24). This results in a complex pattern of tissular, cellular, and molecular remodeling (25), highly variable within an experimental group depending on the scar size. On the other hand, MI is a very useful model to test new therapies aimed at preventing HF. For instance, it has recently been demonstrated that blocking the calmodulin kinase II (CaMKII) can substantially prevent maladaptive remodeling post-MI (see Ca2+ Calmodulin-Dependent Protein Kinase IL).
Ca2+/Calmodulin-Dependent Protein Kinase II)
Because adult cardiac myocytes are terminally differentiated cells that have lost most of their capacity to undergo complete cell division, cardiac hypertrophy is characterized by myocyte hypertrophy associated with hyperplasia of the nonmuscle cells (26). Indeed, if LV dysfunction is not present and the ventricle is hemodynamically compensated, myocyte hypertrophy is the predominant form of cell growth and myocyte proliferation (which is very low) is not significantly greater than that in control hearts (27). During LV concentric hypertrophy, myocyte growth is characterized by parallel addition of sarcomeres that induces lateral growth of the cell. This contrasts with volume overload during which myocytes increase in length due to the addition of sarcomeres in series, and points to the existence of different triggers and signaling pathways between the two types of LVH (28).
Although it is usual to contrast mechanical to neurohumoral triggers of LVH, in reality these two components probably cooperate in most models of chronic hemodynamic overload. This is all the more evident due to the existence of an active RAS in the heart (29), since it has been established that mechanical signals such as myocardial and myocyte stretch trigger myocyte hypertrophy through the involvement of many neurohumoral factors (see Neurohumoral Inhibitors). Nonetheless, recent data have clearly shown that, in contrast to what was postulated earlier (30), myocyte stretch can trigger myocyte hypertrophy directly through the involvement of integrins, myocyte costamere, and cytoskeleton, without the apparent contribution of neurohormones (19,31).
The question of triggers and pathways leading to cardiac myocyte hypertrophy is best understood in its historical context. This approach stresses the importance of cell culture, pioneered by the work of Simpson (32) in demonstrating the prohypertrophic effect of norepinephrine and in identifying, during the 1980s, the vast number of neurohumoral agents, growth factors, and cytokines associated with inducing hypertrophy of isolated neonatal or adult cardiac myocytes. Although isolated myocytes and cell culture systems remain invaluable tools for specific purposes (e.g., in the context of in vivo animal models (19,31)], the technology was surpassed around the mid-1990s by transgenesis, which most often permits similar questions to be addressed in the invaluable in vivo setting. Therefore, this chapter will focus on in vivo and, especially, transgenic models.
Mechanical Initiators
In the context of PO-induced LVH, the existence of a mechanical trigger is obvious. However, identification of the precise trigger responsible for LVH in this setting is not as easy. According to the hypothesis that concentric LVH is aimed at normalizing systolic wall stress, the trigger would be the increased systolic load imposed on cardiac myocytes during their contraction. However, the scientific proof of such a trigger remains elusive since such a pure systolic overload is difficult to reproduce in vitro as well as in experimental models. In fact, most in vitro and in vivo studies have focused on the role of passive (diastolic) stretch. We have shown that following AAC in the rat, hypertension and the associated LVH are delayed and preceded by a phase of volume overload due to RAS activation, which triggers gene reprogramming (most probably through the volume overload-induced stretch of the LV wall) (12). Such a mechanism is probably also involved when a tight TAC abruptly increases LV afterload, resulting in decreased ejection fraction and increased end-systolic volume that stretches the LV both during diastole and systole. It should be emphasized that most experimental models poorly reproduce what actually takes place in humans, especially in the setting of genetic hypertension. However, these models have been invaluable for identifying the triggers and pathways of LVH and progression to HF.
Myocyte Stretch
As discussed previously, whether the stretch signal can be transmitted to the nucleus and genes without the intervention of a neurohumoral signal has been questioned for a long time, especially since the study by Sadoshima et al. that demonstrated that growth of neonatal rat ventricular myocytes (NRVM) following stretch necessitated the autocrine activation of AT1 receptors following the release of angiotensin II (Ang II) (30). At the same time, Yamazaki et al. showed that myocyte stretch induces cardiac myocyte hypertrophy through a mechanism implying, at least in part, an increased synthesis and secretion of Ang II and endothelin-I (ET-1) (33,34). However, a large amount of data has accumulated during the past few years to suggest that stimulation of Ang II receptors by the peptide is not a mandatory step in the cascade of events linking myocyte stretch to gene reprogramming. For instance, the Rho family of small
G-proteins plays a critical role in mediating hypertrophic responses to myocyte stretch, independent of Ang II stimulation (35). Moreover, AT1 receptors can be activated by stretch through an Ang II-independent mechanism (36).
G-proteins plays a critical role in mediating hypertrophic responses to myocyte stretch, independent of Ang II stimulation (35). Moreover, AT1 receptors can be activated by stretch through an Ang II-independent mechanism (36).
Reactive oxygen species (ROS)-mediated extracellular signal-regulated kinase (ERK)1/2 activation also seems to be involved in cardiac myocyte hypertrophy induced by cyclic stretch of low amplitude (5%), and ROS production is greatly stimulated by Ang II-mediated NAD(P)H stimulation. In contrast, cyclic stretch of high amplitude (25%), which activates both extracellular signal-regulated kinase 1/2 and c-Jun N-terminal kinase (JNK), is associated with myocyte apoptosis (37) (Table 3-1).
Another important activation of the upstream mechanism that may be involved in stretch-induced cardiac myocyte hypertrophy is the Na+-H+ exchanger Gene expression and activity of the Na+-H+ exchanger is increased in stretched cardiac myocytes (38) as well as in several models of LVH, including PO (39). The resulting increase in intracellular Na+ concentration decreases the transmembrane Na+ gradient, in turn promoting a rise in intracellular Ca2+ concentration via decreased Na+-Ca2+ exchange. The resulting increase in intracellular Ca2+ may activate a number of signaling pathways, including calcineurin-NFAT, CaMK, PKC, and MAPK cascades, thus providing mechanisms by which the Na+-Ca2+ exchanger can promote hypertrophy. Accordingly, HOE 694, an inhibitor of the exchanger, markedly attenuates stretch-induced activation of Raf-1 and MAPK and increase in protein synthesis, although this compound has no effect on Ang II- and ET-1-induced Raf-1 and MAPK activation (38). NHE inhibition is also able to attenuate LVH and fibrosis in spontaneously hypertensive rats (SHRs) (40) and the cardiomyopathic phenotype and, especially, LV dysfunction and fibrosis in mice overexpressing the β1-adrenergic receptor (41).
Regardless of what the upstream transducer is, a number of downstream effectors such as ERK1/2, p90RSK, p38 kinase (p38), Src, and BMK1 are activated by acute myocyte stretch as well as during chronic PO (42). A recent study indicates that PKC-α and -δ are important regulators in mediating activation of Rho GTPases and MAPKs in the cyclic stretch-induced hypertrophic process (43). However, it must be pointed out that most of these studies have been performed using myocytes isolated from neonatal ventricles, thus raising doubts about the possibility of directly extrapolating the results to adult myocytes and the in vivo setting.
Despite the interest of the aforementioned studies, it is clear that a conceptual leap has been accomplished with the recent identification of important transducers of stretch signaling. Mice deleted of the muscle Lim protein (MLP) gene (a gene that encodes a protein of the Z band of the sarcomere that associates with titin and T-cap/telethonin) exhibit dilated cardiomyopathy (DCM) with no hypertrophy when submitted to PO (31). Myocytes isolated from the ventricle of these mice are unable to express FGP when stretched, whereas they are fully responsive to phenylePhrine (PE) or ET-1 stimulation. Similarly, mice deficient for melusin, a muscle-specific protein that interacts with the integrin β1 cytoplasmic domain, exhibit DCM with no LVH when submitted to PO, whereas LVH in these mice is Perfectly induced by chronic administration of Ang II or PE at doses that do not increase blood pressure (19).
Extra- and Intra-Cellular Signaling
The previously-mentioned studies have generated a renewal of interest in the signals mediated through the cell membrane via the integrins and associated proteins. Integrins are a large family of heterodimeric cell surface receptors that link the extracellular matrix to the intracellular cytoskeleton and signal bidirectionally across the cell membrane of cardiac myocytes and fibroblasts. Most interestingly from the cardiovascular viewpoint, they have been shown to function as mechanotransducers. The repertoire of integrins present on cells is modulated in the remodeled myocardium. Furthermore, hemodynamic loading may alter key integrin-mediated signaling events (44,45). The importance of integrins in the hypertrophic process is strengthened by the observation that β1 integrin knockout mice are intolerant to TAC (46). Other important proteins involved in extra-and intra-cellular signaling are discoidin domain receptors (DDRs), which are transmembrane complexes that attach specifically to collagen which have tyrosine kinase motifs in the cytoplasmic domain, and the cadherins, which regulate cell-cell interaction. The arrangement and localization of these proteins on the surface of myocytes and fibroblasts suggest that they may play an important role not only in force transmission but also in cell signaling (45).
It is important to note that cardiac fibroblasts also have the ability to sense, integrate, and functionally respond to mechanical stimuli through the secretion of autocrine/paracrine factors such as Ang. II, ET-1, or tumor necrisos factor-α (TNF-α) that are released in response to mechanical stimulation and exert their effect on both fibroblasts and cardiac myocytes (45). These neurohumoral factors and cytokines activate intracellular pathways through their binding to their respective receptors. For instance, osteopontin (OPN), a protein secreted by both fibroblasts and cardiac myocytes that binds to integrins, also seems to play an important role in LVH. It plays an important role in cardiac fibrosis, probably through the modulation of cellular adhesion and proliferation of fibroblasts. Its expression is increased in cardiac hypertrophy and its absence attenuates fibrosis (47). Moreover, increased phosphorylation of JNK, p38, Akt, and GSK-3β is significantly lower in the heart of OPN knockout mice submitted to TAC than in their wild type (WT) controls, suggesting that increased OPN expression may play an important role in modulating PO-induced LVH (48).
Neurohumoral Initiators
Catecholamines
Intuitively, the increase in sympathetic tone and activation of the RAS that occurs at the acute phase of MI should play an important role in the subsequent hypertrophy of the noninfarcted myocardial area. It is less clear whether α- and/or β-adrenoceptor activation should play an important role in the development of PO-induced LVH. As previously mentioned, the prohypertrophic effect of α-adrenergic stimulation was the first demonstrated prohypertrophic effect of a neurohumoral agent on isolated cardiac myocytes (32). A similar observation was done for β-adrenoceptor
stimulation (49). The role of α- and β-adrenergic signaling during cardiac hypertrophy and failure has been reviewed in detail recently (50) and will not be detailed here, with the exception of the downstream signaling cascades involved (see Gg/G11 Signaling and Gs/Gi signaling). There are numerous studies of transgenic mice demonstrating both the requirement of norepinephrine and epinephrine for the induction of in vivo PO-induced LVH and the detrimental effects of such an activation; the LVH associated with activation of these receptors and downstream pathways most often results in maladaptive hypertrophy progressing to HF. More interesting is the demonstration of an apparently almost universal requirement of endogenous catecholamines for the induction of in vivo PO-induced cardiac hypertrophy and for the activation of hypertrophic signaling pathways (51).
stimulation (49). The role of α- and β-adrenergic signaling during cardiac hypertrophy and failure has been reviewed in detail recently (50) and will not be detailed here, with the exception of the downstream signaling cascades involved (see Gg/G11 Signaling and Gs/Gi signaling). There are numerous studies of transgenic mice demonstrating both the requirement of norepinephrine and epinephrine for the induction of in vivo PO-induced LVH and the detrimental effects of such an activation; the LVH associated with activation of these receptors and downstream pathways most often results in maladaptive hypertrophy progressing to HF. More interesting is the demonstration of an apparently almost universal requirement of endogenous catecholamines for the induction of in vivo PO-induced cardiac hypertrophy and for the activation of hypertrophic signaling pathways (51).
Table 3-1 Signaling Mechanisms that Initiate Myocyte/Myocardial Remodeling In Vitro and/or In Vivo | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Angiotensin II and Endothelin-1
As already mentioned, Ang II of systemic or tissular origin is probably the most important humoral agent involved in LVH, especially its maladaptive aspects. Ang II acts through binding to its specific receptors, the type 1 and type 2 Ang II receptors (AT1R and AT2R, respectively). AT1R is probably the most important receptor involved in LVH induced by mechanical and/or neurohumoral stimulation. Its level of expression is also influenced by PO. In the long term, AT1R protein and mRNA levels have been found to be consistently increased in all models of PO-induced LVH in the rat (52). However, although subpressor doses of Ang II failed to increase LV mass in mice deleted of the AT1R, PO fully induced LVH in these mice with myocyte hypertrophy, myocardial fibrosis, and expression of FGP, suggesting that AT1-mediated Ang II signaling is not necessary for the development of PO-induced LVH (53,54).
Despite its demonstrated involvement in cardiac development, the role of AT2R in LVH has remained elusive until recently. More precisely, many in vitro and in vivo studies have suggested that AT2R stimulation exerts counterbalancing suppressant effects against the growth-promoting, profibrotic, and hypertrophic effects of AT1R stimulation (55). For instance, lentivirus-mediated overexpression of AT2R in the heart of 5-day-old SHRs led to decreased LV wall thickness and decreased heart weight-to-body weight ratio (HW/BW), indicating that early AT2R overexpression attenuates the development of cardiac hypertrophy in this context (56).
Recent data, however, have suggested a possible different role in which AT2R would support rather than oppose the effects of AT1R stimulation (55). For instance, targeted deletion of mouse AT2R prevented AAC-induced LVH (57), and LVH and fibrosis induced by infusion of a hypertensive dose of Ang II were eliminated in mice lacking the AT2R, indicating that this receptor is essential for LVH and fibrosis in chronic, Ang II-induced hypertension (58). In the same experimental setting in rats, AT2R blockade with PD123319 had no influence on LVH, α-skeletal actin, and β-MHC expression but did increase the expression of c-fos, ET-1, IGF-1, and atrial natriuretic peptides and decreased vascular endothelial growth factor (VEGF) and fibroblast growth factor-1 (FGF-1) expression. This suggests that AT2R plays a functional role in the hypertrophic process by selectively regulating the expression of growth-promoting and growth-inhibiting factors (59).
Endothelin-1 (ET-1) has long been known to be a possible mediator of hypertrophy, both independently and in response to other growth factors, especially Ang II. Both exposure to Ang II and stretching of NRVM induce ET-1 mRNA and secretion of ET-1 in the culture medium. In these cells, the Ang II-induced increase in protein synthesis is blocked by ET-1 antisense oligonucleotides, suggesting that autocrine/paracrine synthesis of ET-1 is an obligatory intermediate step in Ang II-induced myocyte hypertrophy (60). In addition, stretch-induced activation of MAPK and increase in protein synthesis are inhibited by the ET-1 antagonist BQ123 (61). Co-culture of NRVM with cardiac fibroblasts has enabled the demonstration of the interaction between the two cell types, with Ang II stimulating cardiac myocyte hypertrophy, at least in part, via paracrine release of TGF-β1 and ET-1 from fibroblasts (62).
Mineralocorticoid
The role of aldosterone and mineralocorticoid receptor (MR) stimulation, independent of the role of Ang II, in the development of LVH has been a matter of debate. Indeed, if mineralocorticoids are used to induce hypertension and LVH in the deoxycorticosterone acetate (DOCA)-salt model, the actual role of mineralocorticoids on myocyte hypertrophy is unclear; aldosterone is better-known for its profibrotic effects than as an important mediator of myocyte hypertrophy. In fact, aldosterone induces cardiac fibrosis through an increase of cardiac AT1R receptor levels, thereby potentiating the profibrotic effect of Ang II (63). However, small amounts of aldosterone are also produced in the heart and this production is increased by Ang II, suggesting that the heart contains a steroidogenic system that is regulated similarly to the adrenal one (64).
In rats that are double-transgenic for the human renin and angiotensinogen genes (dTGR) and that develop hypertension, vasculopathy, LVH, and fibrosis, both spitonolactone and the AT1R blocker valsartan prevented death and vasculopathy and reversed LVH, while only valsartan normalized blood pressure. This suggests that aldosterone promotes LVH and fibrosis by a mechanism independent of increased blood pressure (65). Transcription factors AP-1 and nuclear factor kappa B (NF-κB) were activated in dTGR compared with controls. MR blockade downregulated these effectors and reduced Ang II-induced cardiac damage (65). Similarly, mice with cardiac-specific overexpression of 11β hydroxysteroid dehydrogenase type 2 (which converts glucocorticoids to receptor-inactive metabolites, thus allowing aldosterone to occupy and activate MRs) spontaneously develop LVH, fibrosis, and HF—all effects mitigated by the MR blocker eplerenone (66). Mice with cardiac-specific conditional overexpression of the human MR exhibit a high rate of death associated with severe ventricular arrhythmias (67). Such alterations are prevented by spironolactone, suggesting novel opportunities for prevention of arrhythmia-related, sudden cardiac death. Unexpectedly, spironolactone also reversed interstitial fibrosis, attenuated myocyte disarray, and improved diastolic function in the cardiac troponin-T
(cTnT)-Q92 transgenic mouse model of human hypertrophic cardiomyopathy (HCM), suggesting aldosterone as an important pathophysiological link between sarcomeric mutations and cardiac phenotype in HCM (68).
(cTnT)-Q92 transgenic mouse model of human hypertrophic cardiomyopathy (HCM), suggesting aldosterone as an important pathophysiological link between sarcomeric mutations and cardiac phenotype in HCM (68).
The prohypertrophic/profibrotic effects of mineralocorticoids on the heart are likely mediated, at least in part, by stimulation of the calcineurin/NFAT pathway, since FK506 or cyclosporine partially prevented LVH and fibrosis in uninephrectomized rats placed on a 1.0% NaCl diet and treated with aldosterone (69). In vitro data suggest that aldosterone also potentiates the effects of Ang II on ROS generation, EGFR transactivation, and ERK1/2 phosphorylation (70). This may explain, at least in part, why MR blockade decreases end-organ damage in dTGR (65).
Peptide Growth Factors and Receptor Tyrosine Kinases
Receptor tyrosine kinases (RTKs) transmit signals from many peptide growth factors, including fibroblast growth factor (FGF), nerve growth factor (NGF), epidermal growth factor (EGF), platelet-derived growth factor (PDGF), transforming growth factor-β (TGF-β), insulin, and insulin-like growth factor-1 (IGF-1). The mechanism of receptor activation includes ligand-induced dimerization and mutual autophosphorylation on tyrosine residues of the cytoplasmic tail. This allows recruiting signaling molecules to the membrane, where they form transient complexes that transmit signals downstream in the cell.
TGF-β and the RAS play a pivotal role in the development of LVH and HF. Recent studies indicate that Ang II and TGF-β do not act independently from one another but, rather, act as part of a signaling network in order to promote cardiac remodeling (71). As previously described, Ang II stimulates cardiac myocyte hypertrophy through paracrine release of TGF-β1 and ET-1 from fibroblasts (62). Ang II upregulates TGF-β expression via activation of the AT1R in cardiac myocytes and fibroblasts, and induction of this cytokine seems necessary for Ang II-induced cardiac hypertrophy in vivo. In the rat, AAC is associated with early myocardial fibroblast activation (proliferation and phenotype transition to myofibroblasts) followed by myocyte hypertrophy and fibrosis. This is associated with early induction of myocardial TGF-β mRNA expression. An anti-TGF-β neutralizing antibody inhibits fibroblast activation and subsequently prevents collagen mRNA induction and myocardial fibrosis, but not myocyte hypertrophy (72). Whether TGF-β is directly involved in myocyte hypertrophy, fibrosis, or both remained a matter of debate until the development of TG mice. TG mice with elevated levels of activated TGF-β1 in the heart show overt fibrosis only in the atria and an inhibition of ventricular fibroblast DNA synthesis, suggesting that TGF-β1 alone is insufficient to promote ventricular fibrosis (73). Although WT mice subjected to chronic subpressor doses of Ang II showed LVH and impaired LV function, TGF-β1-deficient mice showed no significant changes in LV mass and percent LV fractional shortening. Cardiomyocyte cross-sectional area was also markedly increased in Ang II-treated WT mice but was unchanged in Ang II-treated, TGF-β-deficient mice. Atrial natriuretic peptide (ANP) expression was increased sixfold in Ang II-treated WT, but not in TGF-β1-deficient mice. Taken together, these results indicate that TGF-β1 is an important mediator of the hypertrophic growth response of cardiac myocytes to Ang II in vivo (74).
An important downstream effector of TGF-β is the TGF-β-activated kinase TAK1, a member of the MAP kinase kinase kinase family (see Intracellular Signaling Pathways and Mitogen-Activated Protein Kinases). Indeed, TAC-induced LVH in mice is associated with TGF-β upregulation and a sevenfold increase of TAK1 kinase activity. Moreover, an activating mutation of TAK1 expressed in the myocardium of TG mice is sufficient to produce p38 phosphorylation, LVH, interstitial fibrosis, severe LV dysfunction, induction of FGP, apoptosis, and early mouse lethality (75). Another important downstream mediator of TGF-β is connective tissue growth factor (CTGF). This secreted protein binds to cell surface and possesses a broad spectrum of biological effects, including stimulation of fibroblast proliferation, extracellular matrix (ECM) production, and myocyte hypertrophy (76). CTGF is rapidly upregulated in NRVM exposed to phenylephrine or ET-1, suggesting that this growth factor is involved in cardiac hypertrophy and fibrosis (77).
Whether FGF-2 is involved in the development of PO-induced LVH has also been a matter of debate. In one study, mice lacking FGF-2 developed significantly less LVH in response to TAC than did their WT controls (78). In another study, normotensive mice lacking FGF-2 developed DCM and exhibited impaired cardiac growth in response to 2K1C-induced hypertension (79). In vitro experiments from the same study indicated that FGF-2 of fibroblast origin is a crucial mediator of LVH via autocrine/paracrine actions on cardiac myocytes.
Insulin-like growth factor 1, the main mediator of the growth effects of growth hormone (GH), is expressed in the myocardium in response to PO (80). However, evidence is accumulating to suggest that IGF-1 signaling through the recruitment of PI3K/Akt (see Intracellular Signaling Pathways) is involved in physiological/adaptive LVH seen during exercise conditioning or pregnancy rather than in PO-induced pathological/maladaptive LVH (4,81). However, in the latter context, IGF-1 expression may serve as an important cardioprotective mechanism.
Cardiotrophin-1, Leukemia Inhibitory Factor, and the Interleukin-6 Receptor
Following binding of interleukin-6 (IL-6), leukemia inhibitory factor (LIF), or cardiotrophin-1 (CT-1), the transmembrane receptor gp130 is activated by several mechanisms, including heterodimerization with gp80—a process that rapidly activates the Janus kinase/signal transducer and activator of transcription (JAK/STAT) and the extracellular signal-regulated kinase (ERK) pathways. Ang II induces IL-6, LIF, and CT-1 in cardiac fibroblasts, and these cytokines (particularly LIF and CT-1) activate gp130-linked signaling and contribute to Ang II-induced cardiac myocyte hypertrophy (82). Mice double-transgenic for IL-6 and its receptor develop myocardial hypertrophy (83), and mice with cardiac-specific expression of a dominant-negative mutant of gp130 develop less LVH when submitted to TAC than do their WT controls (84). More interestingly in
the context of PO-induced LVH, mice with deleted gp130 develop severe DCM and HF associated with massive myocyte apoptosis when submitted to TAC, indicating an obligatory activation of the gp130 pathway to prevent myocyte apoptosis and HF following PO of the LV (18).
the context of PO-induced LVH, mice with deleted gp130 develop severe DCM and HF associated with massive myocyte apoptosis when submitted to TAC, indicating an obligatory activation of the gp130 pathway to prevent myocyte apoptosis and HF following PO of the LV (18).
Intracellular Signaling Pathways
Gq/G11 Signaling
α1-adrenergic, Ang II, and ET-1 receptors are coupled to heterotrimeric Gq/G11 proteins that activate phospholipase C (Fig. 3-2). Stimulation of all has been shown to be sufficient to induce hypertrophy of NRVM in culture. The role of G-proteins in this process was clarified during the second half of the 1990s with the development of important TG mouse models (4,85). TG overexpression of these receptors, as well as Gq/G11, results in cardiac hypertrophy, which possibly leads to cardiomyopathy with depressed cardiac function depending on the degree of overexpression of the transgene (15). More interesting are the effects of TAC in mice overexpressing Gαq (17). Although nontransgenic mice developed concentric LVH associated with the typical re-expression of FGP, Gαq-overexpressing mice developed excentric hypertrophy leading to HF with no further increase in FGP expression. This study is of special importance because it indicated for the first time that the sole stimulation of a signaling pathway, without mechanical overload, is able to reproduce the ventricular phenotype produced by PO, thereby demonstrating the importance of Gαq stimulation in the development of PO-induced LVH. Also, because HF occurred rapidly after TAC in TG mice with LVs already exhibiting all the features of PO-induced LVH, the study demonstrated the limits of adaptation due to LVH and re-expression of FGP. Moreover, it was also one of the first studies suggesting that the phenotypical changes resulting from PO/Gαq overstimulation may be maladaptive rather than adaptive.
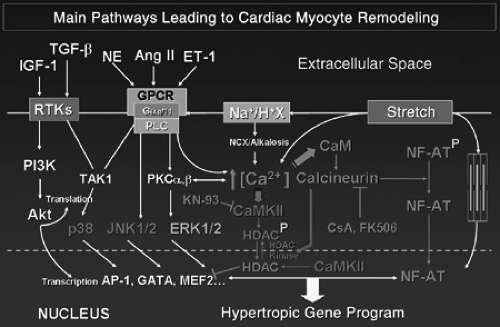 Figure 3-2 Simplified overview of the main pathways leading from biomechanical stress to cardiomyocyte gene reprogramming. Myocyte stretch and stimulation of the Gq protein coupled receptors (GPCR) by norepinephrine (NE), angiotensin II (Ang II) and endothelin (ET-1) are the main upstream initiators of a number of intracellular cascades [MAPK (p38, JNK, ERK), calcineurin-NFAT, CaMK-HDAC, and so forth] whose downstream effectors act on transcription factors and genes to induce the expression of the hypertrophic gene program. Increased cytosolic free calcium concentration plays a central role in this process. On the left, the PI-3K-Akt pathway is the main signaling pathway responsible for physiological LVH resulting from exercise conditioning or pregnancy, with effects on both gene transcription and mRNA translation. |
Conversely, when TAC was performed in mice in which Gq signaling had been blocked by the expression of a specific peptide that uncouples Gq from its coupled receptors, TG mice developed less LVH than did WT controls, further demonstrating the role of Gq in the initiation of myocardial hypertrophy (86). Interestingly, cardiac contractility was preserved in these mice despite the lack of normalization of the LV wall stress, and the animals displayed a slower pace of deterioration of systolic function compared with WT controls. This indicates that LVH is not a mandatory adaptive mechanism, at least in this model and for the time period considered (87). Similarly, in TG mice with cardiac-specific overexpression of RGS4, a protein which inactivates signaling through heterotrimeric G-proteins by increasing the GTPase activity of Gαq/Gαi (88), PO failed to induce LVH (89). Similarly, cross-breeding RGS4-overexpressing mice with Gq-overexpressing mice delayed the LV contractile dysfunction and FGP induction seen in the latter (90). The importance of Gq/G11 signaling in the development of LVH was strengthened by the combined ablation of Gq and G11 (91). Such an approach resulted in an almost complete lack of LVH and activation of FGP in response to TAC, demonstrating the requirement for Gq/G11 for most (if not all) features of PO-induced LVH (91).
An intriguing mechanism has been suggested from in vitro studies that the prohypertrophic effect of norepinephrine, Ang II, and ET-1 would necessitate the activation of the myocardial heparin-binding epidermal growth factor receptor (HB-EGFR), a member of the ErbB family of receptor tyrosine kinases. The process would require the cell surface cleavage and shedding of the ectodomain of HB-EGF by a disintegrin and metalloproteinase (ADAM12), likely activated by PKCδ, which cleaves membrane-bound HB-EGF, allowing HB-EGFR activation in response to GqPCR stimulation or PO (92). In vivo, KBR-7785, an inhibitor of ADAM12, attenuated LVH induced by TAC or PE or Ang II infusion. An antisense oligodesoxynucleotide to EGFR prevented LVH in Ang II-infused rats and attenuated Ang II-enhanced EGFR expression and ERK activation, reinforcing the concept that Ang II requires EGFR to mediate ERK activation in the heart (93). Similar findings were observed with young but not adult SHRs, suggesting a critical role of the EGFR-activated ERK pathway in cardiovascular development but not in the maintenance of established LVH (94).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


