Arrhythmogenic right ventricular cardiomyopathy (ARVC) is an inherited heart muscle disease characterized by fibrofatty replacement of the myocardium and ventricular arrhythmias, associated with mutations in the desmosomal genes. Only a missense mutation in the DES gene coding for desmin, the intermediate filament protein expressed by cardiac and skeletal muscle cells, has been recently associated with ARVC. We screened 91 ARVC index cases (53 negative for mutations in desmosomal genes and an additional 38 carrying desmosomal gene mutations) for DES mutations. Two rare missense variants were identified. The heterozygous p.K241E substitution was detected in 1 patient affected with a severe form of ARVC who also carried the p.T816RfsX10 mutation in plakophilin-2 gene. This DES substitution, showing an allele frequency of <0.01 in the control population, is predicted to cause an intolerant amino acid change in a highly conserved protein domain. Thus, it can be considered a rare variant with a possible modifier effect on the phenotypic expression of the concomitant mutation. The previously known p.A213V substitution was identified in 1 patient with ARVC who was negative for mutations in the desmosomal genes. Because a greater prevalence of p.A213V has been reported in patients with heart dilation than in control subjects, the hypothesis that this rare variant could have an unfavorable effect on cardiac remodeling cannot be ruled out. In conclusion, our data help to establish that, in the absence of skeletal muscle involvement suggestive of a desminopathy, the probability of DES mutations in ARVC is very low. These findings have important implications in the mutation screening strategy for patients with ARVC.
Arrhythmogenic right ventricular cardiomyopathy (ARVC; MIM 107970) is an inherited cardiac disease characterized by progressive fibrofatty myocardial replacement, primarily of the right ventricle. Clinical manifestations occur most often between the second and fourth decade of life and are characterized by ventricular arrhythmias, heart failure, and sudden death. ARVC is the second most common cause of unexpected sudden death among young people and athletes and affects men more frequently than women, with a 3:1 ratio. The disease is familial in about half the cases and typically shows an autosomal dominant inheritance, with reduced penetrance and variable expression. Mutations in the genes encoding several components of the cardiac desmosome are associated with ARVC in about 50% of probands. The presence of multiple mutations in desmosomal genes have been reported in a significant proportion of cases. Nondesmosomal ARVC genes have also been identified, providing evidence that ARVC pathogenesis extends beyond the desmosomes. Currently, only a novel mutation in the DES gene encoding for desmin, the intermediate filament protein expressed by cardiac cells, has been identified in a proband showing a typical form of ARVC. We report the mutation screening of the DES gene in a cohort of Italian patients with ARVC to establish the prevalence of DES mutations.
Methods
We included 91 unrelated patients of Italian descent with ARVC. The clinical evaluation consisted of a detailed personal and family history, physical examination, 12-lead electrocardiogram (ECG), 2-dimensional echocardiogram, signal-averaged ECG, and stress test ECG, performed according to previously reported methods. In all probands, a clinical diagnosis of ARVC was made using the revised 2010 Task Force criteria. None of these patients showed skeletal muscle disease. Available family members of the probands carrying DES variants were also clinically evaluated. All clinical investigations were conducted according to the principles expressed in the Declaration of Helsinki. The ethics committee review board of the University of Padua (Padua, Italy) approved the study protocol. All the participants provided written informed consent before inclusion in the study. Of the 91 patients, 53 were ARVC index cases and were negative for mutations in the desmosomal genes, and 38 patients carried desmosomal gene mutations (11 in the plakophilin-2 [PKP2] gene, 13 in the desmoplakin [DSP] gene, 7 in the desmoglein-2 [DSG2] gene, 2 in the desmocollin-2 [DSC2] gene, and 5 with multiple mutations).
Genomic DNA was isolated from peripheral blood lymphocytes using standard procedures. Primers for polymerase chain reaction amplification of the DES gene were designed using PRIMER3 software (available at: http://frodo.wi.mit.edu/ ; Supplemental Table 1 ). Polymerase chain reaction products were analyzed using denaturing high-performance liquid chromatography and/or direct sequencing. Denaturing high-performance liquid chromatography analysis was performed using the WAVE Nucleic Acid Fragment Analysis System 3500HT equipped with the DNASep HT cartridge (Transgenomic, Omaha, Nebraska). Temperatures for sample analysis were selected using WAVE Navigator software (Transgenomic; Supplemental Table 1 ). Polymerase chain reaction fragments showing an aberrant, temperature-modulated denaturing high-performance liquid chromatography heteroduplex profile were sequenced using the BIG DYE dideoxy-terminator chemistry (PerkinElmer, Waltham, Massachusetts) on an ABI 3730XL DNA sequencer (PE Applied Biosystems, Foster City, California). Chromas, version 1.5, software (Technelysium, South Brisbane, Australia) and Lasergene package computer programs (DNASTAR, Madison, Wisconsin) were used to edit, assemble, and translate the sequence. Numbering of the DES nucleotides starts at ATG and refers to GenBank accession number NM_001927 for the cDNA; the translation sequence was compared to the reference sequence NP_001918. A control group of 300 healthy and unrelated subjects (600 alleles) from the Italian population was used to exclude that novel variants could be common DNA polymorphisms. All the controls were matched to the probands by ancestry. The amplification refractory mutation system was used to screen the control population for the presence of nucleotide changes identified in the DES gene (c.638C>T, c.721A>G, and c.736-11A>G). The primer sequence, amplicon size, and amplification annealing temperatures are reported in Supplemental Table 2 . Mutation screening was performed for all available family members of index cases in which a DES variant was detected.
Results
A systematic mutation screening in the DES coding regions was performed in 53 ARVC unrelated index cases negative for mutations in the desmosomal genes. Because the occurrence of multiple mutations in ARVC has been reported, the DNA of another 38 consecutive and unrelated patients with desmosomal gene mutations (11 in the PKP2 gene, 13 in the DSP gene, 7 in the DSG2 gene, 2 in the DSC2 gene, and 5 carrying multiple mutations) was investigated for DES mutations.
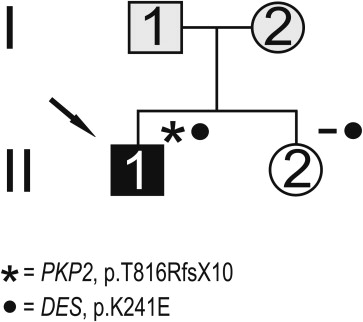
Mutation analysis resulted in the identification of several nucleotide substitutions ( Table 1 ). A heterozygous nucleotide change, c.721A>G, resulting in the p.K241E substitution, was identified in 1 patient ( Figure 1 ). It was not detected in 600 control chromosomes, in single nucleotide polymorphism database (dbSNP; available at: http://www.ncbi.nlm.nih.gov/projects/SNP/ ), or in the Exome Variant Server (available at: http://evs.gs.washington.edu/EVS/ ). Very recently, it was reported in the 1000 Genomes Project database (available at: http://www.1000genomes.org ) with a minor allele frequency of <0.01 (1000GENOMES_2_220285054). This nonconservative amino acid substitution, which was considered intolerant by Condel (available at: http://bg.upf.edu/condel/analysis ), SIFT (available at: http://sift.jcvi.org/ ) and PolyPhen-2 (available at: http://genetics.bwh.harvard.edu/pph2/ ) analysis, involves a highly conserved residue in the 1B α-helix subdomain ( Figure 1 ). The index case carrying the p.K241E variant had no family history of juvenile sudden death, ARVC, or skeletal muscle disease. He was diagnosed at 25 years old after a sustained ventricular tachycardia. The basal ECG showed the presence of negative T waves in leads V 1 to V 5 in the absence of complete right bundle branch block (QRS ≥120 ms). The 2-dimensional echocardiogram showed severe dilation of the right ventricle (RV) with marked hypokinesia of the free wall, reduced RV ejection fraction, and mild dilation of the left ventricle. He was affected by a severe form of ARVC and received an implantable cardioverter defibrillator at 34 years old. He was found to carry a pathogenic frame shift mutation (p.T816RfsX10) in PKP2 gene. The patient’s sister, carrying only the same DES variant, was fully asymptomatic clinically and did not show any cardiac abnormality on the echocardiogram, ECG, or magnetic resonance imaging scan ( Figure 2 ).
| Exon | Nucleotide Change | Amino Acid Change | dbSNP/1000Genomes ID | MAF |
|---|---|---|---|---|
| 1 | c.75A>G | p.P25P | rs1318299 | 0.089 |
| 1 | c.93T>C | p.S31S | rs2017800 | 0.100 |
| 1 | c.408C>T | p.L136L | rs111828114 | 0.021 |
| Intron 1 | c.579-38C>T | − | rs12991025 | 0.456 |
| 2 | c.638C>T | p.A213V | rs41272699 | 0.008; 0.012 ∗ |
| 3 | c.669T>C | p.I223I | rs75882680 | 0.022 |
| 3 | c.721A>G | p.K241E | 1000GENOMES_2_220285054 | <0.01; 0.000 ∗ |
| Intron 3 | c.735+20C>T | − | rs151226355 | 0.005; 0.013 ∗ |
| Intron 3 | c.736-35C>A | − | rs41272701 | 0.018 |
| Intron 3 | c.736-11A>G | − | novel (GenBank JX114779) | 0.000 ∗ |
| 4 | c.828C>T | p.D276D | rs1058261 | 0.347 |
| 5 | c.1014G>C | p.L338L | rs12920 | 0.347 |
| 6 | c.1104G>A | p.A368A | rs1058284 | 0.342 |
∗ MAF estimated in the Italian control population for nucleotide changes not reported in SNP databases or showing MAF <0.01.
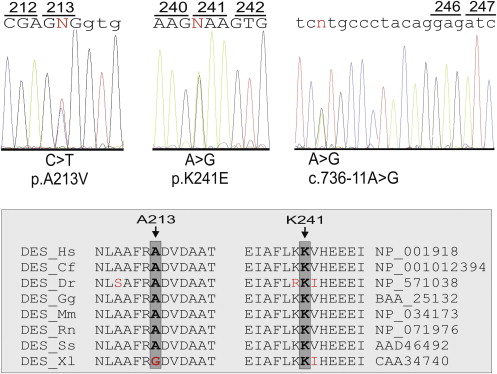
The known c.638C>T nucleotide substitution, resulting in a p.A213V amino acid change, was identified in 1 patient who was negative for mutations in the desmosomal ARVC genes ( Figure 1 ). This variant currently appears in single nucleotide polymorphism database (rs41272699, minor allele frequency 0.8%). In the present study, it was detected in the healthy controls at an allele frequency of 1.2%. The index patient carrying the p.A213V variant was diagnosed at 47 years old after a presyncopal episode. The basal ECG showed the presence of negative T waves in leads V 1 to V 3 in the absence of complete right bundle branch block (QRS ≥120 ms). Echocardiography showed mild RV dilation with kinetic abnormalities in the RV free wall. Magnetic resonance imaging confirmed the presence of mild dilation of the right ventricle, together with kinetic alterations of the RV free wall, and showed adipose tissue infiltration along the RV free wall. No family history of juvenile sudden death, ARVC, or skeletal muscle disease was reported. No family members were available for clinical and genetic testing.
A heterozygous nucleotide change close to the acceptor splice site of exon 4 (c.736-11A>G) was identified in 1 patient in whom no mutation was detected in the desmosomal ARVC genes ( Figure 1 ). He was examined at 46 years old because of effort dyspnea. The basal ECG showed the presence of negative T waves in leads V 1 to V 3 in the absence of complete right bundle branch block and an epsilon wave in the right precordial leads. The 2-dimensional echocardiogram revealed the presence of severe RV dilation, depression of the RV fractional area change, apical and subtricuspid akinesia, and apical sacculations. The detected unclassified variant, which was absent in 600 control chromosomes and in the above-mentioned databases, was identified in 2 of 5 affected and 3 unaffected members of the family. No skeletal muscle and cardiac biopsy samples were available to perform additional studies to check for perturbed splicing of the DES mRNA transcript. On the basis of the absence of co-segregation with the disease phenotype and the identification in unaffected family members, this intronic variant was unlikely the causal mutation in this family.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


