Cytoskeletal Abnormalities in Cardiocytes During Heart Failure
George Cooper, IV
The cytoskeleton as classically defined for eukaryotic cells consists of three systems of protein filaments. These are the microtubules, the intermediate filaments, and the microfilaments. In mature striated muscle such as the heart of the adult mammal, these three types of cytoskeletal filaments are superimposed spatially on the specialized system of contractile protein filaments, the myofilaments. A very useful classification of these and the other structural proteins of the cardiac muscle cell has been provided by Schaper et al. in terms of five families of structurally related proteins (1), and an equally useful review of the functional significance of each class of proteins has been provided by White et al. (2). The first of these groupings consists of the contractile proteins of the sarcomere, actin and myosin, and their regulatory proteins, including the troponins and tropomyosin. The second of these groupings consists of noncontractile sarcomeric structural proteins such as α-actinin and titin. The third group consists of membrane-associated proteins such as dystrophin, vinculin, talin, and spectrin. The fourth group comprises the proteins of the intercalated disk, of the adheren junctions, and of the gap junctions. The fifth group, which is the subject of this chapter, consists of the classical cytoskeletal proteins. These are tubulin, the predominant protein of the microtubules; desmin, the predominant protein of the intermediate filaments in muscle; and actin, the predominant protein of the microfilaments. As a general statement, the protein filaments that comprise the classical cytoskeleton interrelate the other four groupings of structural proteins as well as the cellular organelles and thus maintain the structural and functional integrity of the cardiac muscle cell.
During the progression from cardiac normality through compensated cardiac hypertrophy to decompensated cardiac failure, there are a number of changes within the cardiac muscle cell, or cardiocyte, in the microtubule network and in the intermediate filament network, without apparent changes in the microfilaments (3,4). For the microtubules, the changes are solely secondary (i.e., they occur only in response to imposed pathological challenges) (5,6,7,8). For the intermediate filaments, there are some similarly reactive secondary changes (7,8,9,10), but there are also primary abnormalities of the intermediate filaments which can themselves lead to cardiac failure (11,12,13). For the microfilaments, neither primary nor specific secondary changes have been described in response to or causative of cardiac hypertrophy and failure (1).
Microtubules
Microtubules, which are hollow protein cylinders about 25 nm in diameter, are composed primarily of α-tubulin and β-tubulin. Each of these proteins is encoded by a multigene family and they form the αβ-tubulin heterodimer immediately after protein translation (14). These heterodimers form filaments that nucleate in the cardiocyte along the nuclear membranes (the minus end of the microtubule) and then extend outward toward the cell periphery (the plus end of the microtubule) (15). They are quite dynamic-constantly undergoing cycles of polymerization, abrupt depolymerization, and repolymerization (16).
In the mature cardiocyte, where the mitotic spindle does not form since cell division has ceased, the normal role for the microtubules is to subserve intracellular trafficking (17,18,19). Here, the microtubule network can be thought of as a railroad yard within which locomotives pull cargos
along microtubule tracks. There are two classes of these locomotives. The first of these is the dyneins, which move cargos along microtubules toward their minus end (i.e., from the cell periphery toward the cell center). The second class is the kinesins, which move cargos along microtubules toward their plus end (i.e., from the center of the cell toward the periphery). Given the fact that the mature striated muscle cell is packed with structural proteins, it is a very diffusion-restricted space, such that long-range movement of macromolecules and particles within these very large cells is virtually impossible. Because of this, movement of membrane vesicles, organelles, and macromolecular complexes by these motor proteins along microtubule tracks is essential to cellular homeostasis. Examples of how this railway system operates include the movement of membrane vesicles to enable the recycling of activated G protein-coupled receptors (18,19) and the movement of mRNA (20) as a component of ribonucleoprotein particles to sites of structural protein synthesis such as the sarcomere.
along microtubule tracks. There are two classes of these locomotives. The first of these is the dyneins, which move cargos along microtubules toward their minus end (i.e., from the cell periphery toward the cell center). The second class is the kinesins, which move cargos along microtubules toward their plus end (i.e., from the center of the cell toward the periphery). Given the fact that the mature striated muscle cell is packed with structural proteins, it is a very diffusion-restricted space, such that long-range movement of macromolecules and particles within these very large cells is virtually impossible. Because of this, movement of membrane vesicles, organelles, and macromolecular complexes by these motor proteins along microtubule tracks is essential to cellular homeostasis. Examples of how this railway system operates include the movement of membrane vesicles to enable the recycling of activated G protein-coupled receptors (18,19) and the movement of mRNA (20) as a component of ribonucleoprotein particles to sites of structural protein synthesis such as the sarcomere.
Microtubules in Cardiac Pathology
As indicated earlier, there are no reports of primary microtubule defects that are responsible for cardiac pathology. This may be due to the fact that the isoforms comprising each member of the tubulin heterodimer pair are encoded by one or more members of a multigene family rather than by alternative splicing of a single gene (14). Thus, there is much more plasticity and redundancy for the expression of these proteins than would be the case were each of them encoded by a single gene (see the section on desmin, later). Indeed, different isoforms of β-tubulin are expressed in the heart developmentally and there is no evidence that these isoforms differ functionally in mammals (21).
There are, however, alterations of the microtubule portion of the cytoskeleton that occur secondary to patho-physiological insults to the heart and that may contribute to a poor outcome. In some cases described later for animal models of human disease, this may take the form of a vicious cycle wherein changes in the cardiocyte microtubule network may interfere with the compensatory response of the heart to pathological challenges in terms of both contractile function and hypertrophic growth.
Cardiac Hypertrophy and Failure Induced by Ventricular Pressure Overloading
An association between pressure overload cardiac hypertrophy and densification of the cardiocyte microtubule network was initially reported as a transient change affecting a minority of the cardiocytes from the overloaded ventricle during the early stages of this growth process (16). An increase in cardiocyte microtubules was later found to be a persistent feature of cardiac hypertrophy in response to a substantial pressure overload that affected all of the cardiocytes in the overloaded ventricle (3,4); this cytoskeletal change was associated with contractile dysfunction and the eventual development of congestive heart failure (22,23). This finding in right ventricular myocardium was not present at any stage of the hypertrophic process for an equivalent degree and duration of right ventricular hypertrophy in response to a volume overload that did not result in cardiac dysfunction (3,24). Taken together, these findings suggested that for hemodynamic overloads microtubule network densification is a persistent and relatively specific feature of the cardiac cytoskeletal response to severe pressure overloading. Of most interest, however, was the fact that depolymerization of this dense microtubule network, which imposes a viscous load on the contracting myofilaments (25), restored the initially abnormal cellular contractile function characteristic of severe right ventricular pressure overload hypertrophy to normal (3,4,22,26).
In attributing the improvement in contractile function to removal of this viscous load, it was important to be sure that this effect was not due to unrecognized side effects of colchicine, the drug used for this purpose, or the resultant increased concentration of αβ-tubulin heterodimers, since there are data to suggest that this heterodimer may act as a functional analog of G proteins to activate adenyl cyclase and increase calcium transients when the αβ-tubulin concentration is increased by colchicine (27). However, the restoration of normal contractile function was seen regardless of whether the microtubules were depolymerized using the chemical agent colchicine or, instead, by using the physical modality of transient hypothermia (3,4,28); colchicine did not augment either cardiocyte cyclic adenosine monophosphate (cAMP) levels or their calcium transients (29). Furthermore, microtubule depolymerization via vincristine, which decreases the soluble αβ-tubulin heterodimer concentration, duplicates the mechanical effects on cardiocytes of microtubule depolymerization via colchicine, which increases the soluble αβ-tubulin heterodimer concentration (30). Thus, a positive inotropic effect of αβ-tubulin heterodimers (independent of the direct physical consequences of microtubule depolymerization) was excluded as an explanation for the ameliorative effects of this intervention on the contractile dysfunction of pressure-hypertrophied myocardium.
In the clinically more important setting of left ventricular pressure overload hypertrophy, the findings were very similar to those for right ventricular hypertrophy, but it was also possible to assign more specificity to the setting in which the cytoskeletal abnormality occurs (23). Thus, Figure 4-1 shows that when mongrel dogs of random sex are submitted to a gradually increasing pressure overload on the left ventricle, with aortic pressure gradients as high as 180 mm Hg, the animals tend to segregate into two groups in terms of the extent of compensatory hypertrophy which is produced. Figure 4-2 shows that this has clear consequences in terms of left ventricular function. In the “Hypertrophy” group of dogs (which had compensated left ventricular hypertrophy), the relationship between mean normalized systolic ejection rate and mean systolic stress remained normal throughout the hypertrophic process. In the “Failure” group of dogs (which developed left ventricular failure), there was a progressive reduction in left ventricular function during the process of noncompensatory hypertrophy and, at the time of final study, there was a very significant increase in left ventricular wall stress.
A comparison of the feline right ventricular pressure overload model to the canine left ventricular pressure overload model provided an initial insight into the specific setting in which hemodynamically driven microtubule
network densification occurs. In the right ventricular pressure overload hypertrophy and failure model there is a uniform increase in microtubule network density in all cardiocytes from these ventricles; as discussed elsewhere (5), it is likely but not known that these right ventricles had high wall stress from the onset of pressure overloading. Indeed, Figure 4-3 shows that increased tubulin polymer and total tubulin, as well as microtubule network densification, are confined to failing left ventricles where high wall stress can be accurately documented in the intact animal. Figure 4-2 shows that this has a clear functional consequence at the level of ventricular contraction, and Figure 4-4 shows that this has an equally clear functional consequence at the level of sarcomere contraction in the cells from these same ventricles. That is, in the animals with compensated left ventricular hypertrophy, contractile function at the level of the cardiocyte sarcomeres is well-preserved both at the midpoint of the study when a biopsy was done and at the end of the study. However, in the animals with decompensated left ventricular hypertrophy, while sarcomere contractile function was normal at the midpoint of the study when the biopsy was taken, it had become distinctly abnormal at the end of the study when wall stress was very high. However, Figure 4-4D shows that even in cardiocytes from failing left ventricles, microtubule depolymerization restores grossly abnormal sarcomere contractile function to normal, strongly suggesting that microtubule network densification has an important role to play in the contractile dysfunction seen in this particular form of hypertrophy. Thus, the phenomenon of microtubule network densification with an associated contractile dysfunction apparently specific to this cytoskeletal alteration was originally observed in the pressure overloaded right ventricle. Figures 4-1, 4-2, 4-3, 4-4 confirm this finding in the pressure overloaded left ventricle but assign much more specificity to this pathophysiology in that the linked cytoskeletal and contractile abnormalities are found only with severe pressure overloaded hypertrophy wherein the hypertrophic response had not been sufficient to maintain normal left ventricular wall stress.
network densification occurs. In the right ventricular pressure overload hypertrophy and failure model there is a uniform increase in microtubule network density in all cardiocytes from these ventricles; as discussed elsewhere (5), it is likely but not known that these right ventricles had high wall stress from the onset of pressure overloading. Indeed, Figure 4-3 shows that increased tubulin polymer and total tubulin, as well as microtubule network densification, are confined to failing left ventricles where high wall stress can be accurately documented in the intact animal. Figure 4-2 shows that this has a clear functional consequence at the level of ventricular contraction, and Figure 4-4 shows that this has an equally clear functional consequence at the level of sarcomere contraction in the cells from these same ventricles. That is, in the animals with compensated left ventricular hypertrophy, contractile function at the level of the cardiocyte sarcomeres is well-preserved both at the midpoint of the study when a biopsy was done and at the end of the study. However, in the animals with decompensated left ventricular hypertrophy, while sarcomere contractile function was normal at the midpoint of the study when the biopsy was taken, it had become distinctly abnormal at the end of the study when wall stress was very high. However, Figure 4-4D shows that even in cardiocytes from failing left ventricles, microtubule depolymerization restores grossly abnormal sarcomere contractile function to normal, strongly suggesting that microtubule network densification has an important role to play in the contractile dysfunction seen in this particular form of hypertrophy. Thus, the phenomenon of microtubule network densification with an associated contractile dysfunction apparently specific to this cytoskeletal alteration was originally observed in the pressure overloaded right ventricle. Figures 4-1, 4-2, 4-3, 4-4 confirm this finding in the pressure overloaded left ventricle but assign much more specificity to this pathophysiology in that the linked cytoskeletal and contractile abnormalities are found only with severe pressure overloaded hypertrophy wherein the hypertrophic response had not been sufficient to maintain normal left ventricular wall stress.
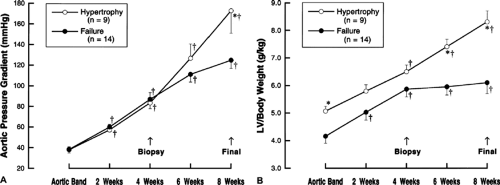 Figure 4-1 Canine left ventricular (LV) afterload increases and resultant LV mass increments with progressive stenosis of the ascending aorta. The group of dogs denoted as “Hypertrophy” maintained normal LV function throughout the course of the study; the group of dogs denoted as “Failure” developed LV systolic dysfunction as the afterload became more severe. Both panels: The times at which the animals were studied and the aortic band was tightened are indicated on the abscissa; the vertical arrows indicate the times of LV biopsy and of final study. A, The aortic pressure gradient as measured by cardiac catheterization is indicated on the ordinate. B, The LV to body weight ratio as measured by ventriculography is indicated on the ordinate. Statistical comparisons, which considered all dogs from a given experimental group together, were by two-way ANOVA and a means comparison contrast, where n = number of dogs. (Reproduced from Tagawa H, Koide M, Sato H, et al. Cytoskeletal role in the transition from compensated to decompensated hypertrophy during adult canine left ventricular pressure overloading. Circ Res. 1998; 82: 751–761 , with permission.)* p <0.01 for difference between the two groups of dogs at matched time points. † p <0.01 for difference from the initial aortic band value within a group. |
When the effect of microtubule depolymerization by colchicine was assessed in vivo, similar results were found at the level of left ventricular function in the intact dog (28) and mouse (30). Figure 4-5A shows that when dogs having the same characteristics as the “Failure” dogs shown in Figure 4-1 were studied in the cardiac catheterization laboratory, they initially showed depressed left ventricular function and elevated left ventricular wall stress, much like that shown for this group of dogs at final study in Figure 4-2. When the aortic pressure gradient was relieved, there was, as expected, a marked reduction in left ventricular wall stress (Fig. 4-5A, 1→2). There was then no immediate effect of intravenous colchicine on left ventricular wall stress or function (Fig. 4-5A, 2→3), since it takes ∼45 minutes for the microtubules to depolymerize after colchicine administration (3). However, 1 hour after intravenous colchicine, when virtually complete depolymerization of the myocardial microtubules had occurred, there was a marked increase in the function of the left ventricle in vivo (Fig. 4-5A, 3→4). This occurred despite the fact that, as
shown in Figure 4-5B, in normal dogs colchicine is, if anything, an intrinsically negative inotropic agent.
shown in Figure 4-5B, in normal dogs colchicine is, if anything, an intrinsically negative inotropic agent.
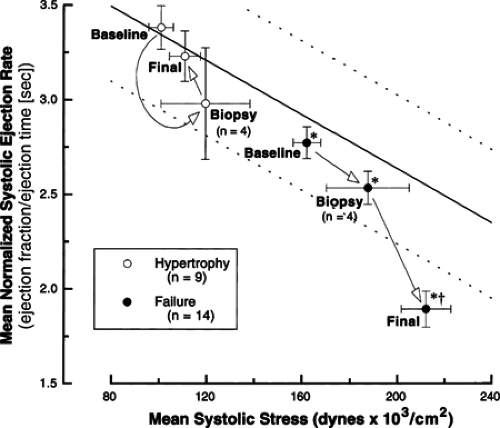 Figure 4-2 The relationship between mean normalized systolic ejection rate and mean systolic stress for the same two groups of dogs whose data are given in Figure 4-1. The solid and parallel dashed lines define this relationship and its 95% confidence interval, calculated using a least-squares linear regression analysis, for 40 β-blocked normal dogs studied in this laboratory. The arrows indicate for each group (separately) the progression through the indicated time points of this study. Statistical comparisons, which considered all dogs from a given experimental group together, were by two-way ANOVA and a means comparison contrast, where n = the number of dogs in each group and the number of dogs in the subset of each group submitted to LV biopsy. (Reproduced from from
Get Clinical Tree app for offline access
Tagawa H, Koide M, Sato H, et al. Cytoskeletal role in the transition from compensated to decompensated hypertrophy during adult canine left ventricular pressure overloading. Circ Res. 1998; 82: 751–761
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree


|