Cytokines as Mediators of Disease Progression in the Failing Heart
Douglas L. Mann
Since the original description of inflammatory cytokines in patients with heart failure in 1990 (1), there has been a growing interest in the role that these molecules play in regulating cardiac structure and function, particularly with regard to their potential role in disease progression in heart failure. The growing appreciation of the pathophysiological consequences of sustained expression of proinflammatory mediators in preclinical and clinical heart failure models culminated in a series of multicenter clinical trials that utilized targeted approaches to neutralize tumor necrosis factor (TNF) in patients with moderate to advanced heart failure. However, these targeted approaches appear to have resulted in worsening heart failure (2), thereby raising a number of important questions about what role, if any, proinflammatory cytokines play in the pathogenesis of heart failure. To this end, in the present review we will summarize the tremendous growth of knowledge that has taken place in this field since 1990, with an emphasis on discussing the adaptive and maladaptive effects of cytokine signaling in the heart, as well as discuss what we have learned from the negative clinical trials that have used targeted anticytokine approaches in heart failure.
The Biology of Proinflammatory (Stress-Activated) Cytokines and their Receptors
The term cytokine is applied to a group of relatively low-molecular-weight protein molecules (generally 15 to 30 Kda) that are secreted by cells in response to a variety of different inducing stimuli. Although cytokines are similar to polypetide hormones in many respects, cytokines can be produced by a variety of different cell types in a number of different tissues, as opposed to being produced by a specific cell type in a specific organ, as is the case for polypeptide hormones. Whereas proinflammatory cytokines have traditionally been thought to be produced by the immune system, one of the more recent intriguing observations is that virtually all nucleated cell types within the myocardium, including cardiac myocytes themselves, are capable of synthesizing a portfolio of proinflammatory cytokines in response to various forms of cardiac injury (Table 13-1). Thus, from a conceptual standpoint, these molecules should be envisioned as proteins that are produced locally within the myocardium by cardiocytes (i.e., cells that reside within the myocardium), in response to one or more different forms of environmental stress. An important corollary of this statement is that the expression of these stress-activated cytokines can occur in the complete absence of activation of the immune system. The biological properties of several important proinflammatory cytokines, including TNF, members of the interleukin-1 (IL-1) family as well as the interleukin-6 (IL-6) family of cytokines, have been reviewed recently and will not be discussed further herein (3).
Adaptive effects of Proinflammatory Cytokine Signaling in the Heart
Although the exact role proinflammatory cytokines play in the heart is not known with certainty, two important themes have emerged from recent studies of proinflammatory
cytokine gene regulation in the heart. One consistent theme is that proinflammatory cytokines are not expressed constitutively in the heart (4,5). The second theme is that these molecules are consistently and rapidly expressed in response to a variety of different forms of myocardial injury (Table 13-1). The observation that proinflammatory cytokine gene expression is not coupled to a specific form of cardiac injury, but is instead observed in all forms of cardiac injury, suggests that these molecules constitute part of an intrinsic or innate stress response system in the heart. Thus, analogous to the role that proinflammatory cytokines play as effector molecules in the innate immune system, which is intended to act as an early warning system that allows the host to rapidly discriminate self from nonself (6), the expression of proinflammatory cytokines in the heart may permit the myocardium to rapidly respond to tissue injury as part of an early warning system that coordinates and integrates a panoply of homeostatic responses within the heart following tissue injury.
cytokine gene regulation in the heart. One consistent theme is that proinflammatory cytokines are not expressed constitutively in the heart (4,5). The second theme is that these molecules are consistently and rapidly expressed in response to a variety of different forms of myocardial injury (Table 13-1). The observation that proinflammatory cytokine gene expression is not coupled to a specific form of cardiac injury, but is instead observed in all forms of cardiac injury, suggests that these molecules constitute part of an intrinsic or innate stress response system in the heart. Thus, analogous to the role that proinflammatory cytokines play as effector molecules in the innate immune system, which is intended to act as an early warning system that allows the host to rapidly discriminate self from nonself (6), the expression of proinflammatory cytokines in the heart may permit the myocardium to rapidly respond to tissue injury as part of an early warning system that coordinates and integrates a panoply of homeostatic responses within the heart following tissue injury.
Table 13-1 Cardiac Pathophysiological Conditions Associated with Activation of Inflammatory Mediators | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
As will be discussed later, there is a now growing body of evidence that supports the point of view that short-term expression of proinflammatory cytokines is beneficial in the heart. However, it bears emphasizing that the family of proinflammatory molecules that comprise this innate stress response system are phylogenetically ancient, and are thus likely evolved in organisms with relatively short life spans (weeks to months). Thus, activation of the innate stress response system was never intended to provide longterm adaptive responses to the host organism. As will be discussed toward the end of this review, sustained and/or dysregulated expression of proinflammatory cytokines is sufficient to produce tissue injury and provoke overt cardiac decompensation.
The first line of evidence in support of a beneficial role for cytokines in the heart is implicit in phylogenetic studies of so-called primitive cytokines, such as TNF and IL-1. TNF and IL-1-like activity has been identified in both protostome vertebrates (annelids) and deuterostome invertebrates (echinoderms) (7,8), suggesting that these molecules came into existence during the onset of the Cambrian period, before the split of the major animal phyla into vertebrate and invertebrate species. The evolutionary development of cytokines was probably necessary for the development of large, multicellular organisms that required intercellular messengers such as cytokines to coordinate complex biological cellular responses. The observation that primitive cytokines such as TNF and IL-1 have been conserved by nature throughout the animal kingdom for nearly 600,000 million years, coupled with the observation that these same cytokines are expressed in virtually all forms of cardiac injury (Table 13-2), suggests that these molecules may in some way confer a survival benefit in the host organism. Nonetheless, this argument is based on teleological evidence and must therefore be regarded as indirect proof in support of the point of view that cytokines confer beneficial responses in the heart.
The second line of evidence in support of a beneficial role for proinflammatory cytokines in the heart comes from a series of gain of function studies that have shown that proinflammatory cytokines confer cytoprotective responses in the heart. The first study to demonstrate the potential beneficial effects of cytokines showed that pretreating rats with TNF protected the heart from ischemic reperfusion injury ex vivo (9). Following ischemia reperfusion injury the hearts from TNF-pretreated animals had a threefold reduction in the amount of lactate dehydrogenase release (9), and showed an increase in the percent of recovery of developed left ventricular pressure when compared to control hearts (10). IL-1 has also been shown to protect rat hearts against ischemia reperfusion injury in vitro (11). Subsequent in vitro studies have demonstrated that physiological levels of TNF are sufficient to protect cardiac myocytes against either hypoxic or ischemic injury, respectively (12). Moreover, the cytoprotective effects of TNF could be mimicked by stimulating either the type 1 (p55, TNFR1) or the type 2 (p75, TNFR2) receptor, suggesting that the cytoprotective effects of TNF are mediated by activation of TNFR1 or TNFR2.
Although these studies did not clearly identify the mechanism for these findings, proinflammatory cytokines are known to upregulate the expression of at least two sets of protective proteins in the heart: the free radical scavenger manganese-superoxide dismutase (MnSOD) (9,13) and the cytoprotective heat shock proteins (HSPs) (14,15). Relevant to this discussion is the finding that TNF-induced MnSOD induction is very rapid (<1 hour) and requires very low levels of TNF (0.1 ng/mL-1), consistent with the proposed homeostatic role for these proteins (13). Given
that contacting myocardial cells are continually susceptible to oxygen-derived free radicals, TNF and IL-1 may play important roles in protecting the heart against oxidative stress, particularly during ischemia and reperfusion injury. TNF has also been shown recently to upregulate the expression of heat shock protein 72 (HSP 72) (14), a protein that is thought to protect the heart against ischemia reperfusion injury (16,17).
that contacting myocardial cells are continually susceptible to oxygen-derived free radicals, TNF and IL-1 may play important roles in protecting the heart against oxidative stress, particularly during ischemia and reperfusion injury. TNF has also been shown recently to upregulate the expression of heat shock protein 72 (HSP 72) (14), a protein that is thought to protect the heart against ischemia reperfusion injury (16,17).
Table 13-2 Deleterious Effects of Inflammatory Mediators in Heart Failure | |||||||||
|---|---|---|---|---|---|---|---|---|---|
|
Finally, proinflammatory cytokines such as TNF and IL-1β have been shown to activate the transcription factor nuclear factor-kappa B (NF-KB), which has been shown to be cytoprotective under certain circumstances, presumably through upregulation of one or more cytoprotective genes, including MnSOD, the cellular inhibitors of apoptosis 1 and 2 (c-IAP1 and cIAP2), and the members of the Bcl-2 family, including Bcl-2, Bcl-1 and Bcl-xL (18,19,20,21,22).
Similar findings have been obtained in gain of function studies for the so-called IL-6 family of cytokines, that include interleukin-6 (IL-6), leukemia inhibitory factor (LIF), cardiotrophin-1 (CT-1), ciliary neurotrophic factor (CNTF), interleukin-11 (IL-11), and oncostatin M (OSM). This family of cytokines triggers downstream signaling pathways in multiple cell types, including cardiac myocytes, either through the homodimerization of the gp130 receptor or through the heterodimerization of gp130 with a related transmembrane receptor. Studies with CT-1 have shown that CT-1 blunts serum deprivation-induced apoptosis in isolated neonatal cardiac myocytes through a pathway that was dependent on activation of the mitogen-activated protein kinase (MAPK). In these studies, transfection of an MAP kinase kinase 1 (MEK1)-dominant negative-mutant cDNA into myocardial cells or treatment with a MEK-specific inhibitor (PD098059) blocked the antiapoptotic effects of CT-1, indicating a requirement of the MAP kinase pathway for the survival effect of CT-1. Similarly, studies have shown that LIF confers cytoprotective responses in isolated myocytes, as well as in intact myocardial tissue (10,23). However, the mechanisms for LIF-mediated cytoprotective effects appear to be more complex than those reported for CT-1. Although studies in isolated neonatal myocytes suggest an important role for the Janus kinase (JAK) and the signal transducer and activator of transcription (STAT)-mediated signaling pathways (23), more recent studies in adult myocytes suggest that the cytoprotective effects of LIF are mediated through activation of the MAPK pathway, consistent with what has been reported for CT-1. One explanation for these apparent differences in the cytoprotective mechanisms for LIF is that there may be functionally significant cross-talk between the MAPK and the JAK/STAT pathways, as has been suggested recently (23). Thus, the cytoprotective signaling pathways that are downstream from gp130-mediated signaling may involve both JAK/STAT- and MAPK-mediated signaling pathways.
The third, and perhaps most striking line of evidence in support of a beneficial role for proinflammatory cytokines in the heart comes from a series of loss of function studies in mice deficient in proinflammatory cytokine receptor-mediated signaling. Mice with targeted disruption of gp130 have been developed recently (24). Mice homozygous for the gp130 knockout (gp130-/-) died between 12.5 days postcoitum and term. The ventricular myocardium in these mice developed normally until 14.5 days postcoitum; however, beyond 16.5 days postcoitum the gp130-/- mice demonstrated a markedly hypoplastic ventricle, with an abnormally thin ventricular wall that had a minimum thickness of one cell. Studies employing mice that harbor a ventricular restricted knockout of the gp130 cytokine receptor via Cre- LoxP-mediated recombination showed that these mice have normal embryonic viability and no evidence of cardiac morphological abnormalities that were observed in the gp130 knockout (gp130-/-) (25). Moreover, these mice had normal cardiac structure (Fig. 13-1B) and function under basal conditions. Thus, the most likely explanation for the findings in the gp130-/- mice is that the cardiac developmental defects and embryonic lethality in these mice were the result of hematopoietic abnormalities and associated oxygen deprivation, as opposed to a primary gp130-mediated defect in myocytes. Interestingly, mice harboring the ventricular restricted knockout of the gp130 cytokine receptor demonstrated a critical role for a gp130-dependent myocyte survival pathway following aortic banding (25). That is, following hemodynamic overloading by transaortic constriction, these mice displayed a decrease in survival (Fig. 13-1A), ventricular enlargement involving both the right and left ventricular chambers (Fig. 13-1B), and a striking increase in the prevalence of cardiac myocyte apoptosis when compared to control mice that exhibited normal compensatory cardiac hypertrophy (25). These studies suggest that the gp130 pathway is an essential stress-activated myocyte survival pathway.
More recently, studies in mice that are doubly deficient for the type 1 and type 2 TNF receptors (TNFR1-/-/TNFR2-/-) have been shown to have an increase in infarct size in response to ischemic injury (26). However, unlike gp130 mice, TNFR1-/-/TNFR2-/- mice displayed a normal cardiac phenotype under nonstressed conditions. However, following acute coronary artery ligation there was a striking increase in infarct size in TNFR1-/-/TNFR2-/- mice (Fig. 13-2A) compared to littermate controls (Fig. 13-2B). As shown by the group data summarized in Figure 13-2C, infarct size was the same in wild-type, TNFR1-/-/TNFR2-/- mice; however, infarct size was 40% greater in the TNFR1-/-/TNFR2-/- mice. Interestingly, the increase in infarct size in the TNFR1-/-/TNFR2-/- mice was shown to be secondary to accelerated apoptosis in the TNFR1-/-//TNFR2-/- mice, as opposed to increased myocyte necrosis. Although this study did not identify the biological mechanisms that were responsible for the cytoprotective effects of TNF, the observation that deletion of both TNFR1 and TNFR2 was necessary to provoke increased tissue injury suggested that TNFR1 and TNFR2 activated redundant cytoprotective signaling pathways in the heart. Although the complete portfolio of cytoprotective signaling pathways that are common to both TNFR1 and TNFR2 is not known, it is interesting to note that NF-KB activation is common to both TNF receptors (27).
As previously noted, NF-KB activation has been shown to be cytoprotective in certain settings. Thus, taken together, the gain of function and loss of function studies for gp130- and TNF-mediated signaling described in this section suggest that proinflammatory cytokines may play an important role in the orchestration and timing of the myocardial stress response, by providing early antiapoptotic
cytoprotective signals that are responsible for delimiting tissue injury, as well as providing delayed signals that facilitate tissue repair and/or tissue remodeling once myocardial tissue damage has supervened. In keeping with this latter point of view, previous studies have shown that CT-1, LIF, and TNF are all sufficient to provoke modest hypertrophic growth response in cardiac myocytes (28) and that TNF is sufficient to lead to degradation and remodeling of the extracellular matrix in the heart (29).
cytoprotective signals that are responsible for delimiting tissue injury, as well as providing delayed signals that facilitate tissue repair and/or tissue remodeling once myocardial tissue damage has supervened. In keeping with this latter point of view, previous studies have shown that CT-1, LIF, and TNF are all sufficient to provoke modest hypertrophic growth response in cardiac myocytes (28) and that TNF is sufficient to lead to degradation and remodeling of the extracellular matrix in the heart (29).
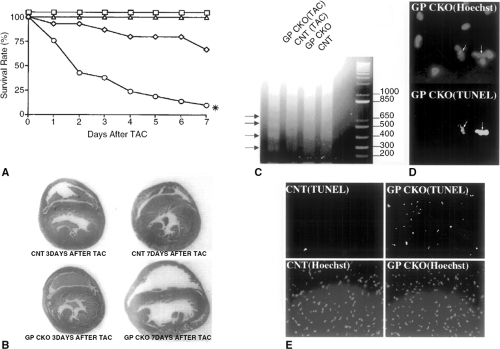 Figure 13-1 Effect of hemodynamic overload in mice with a ventricular restricted knockout of the gp130 signaling pathway. (A) Analysis of survival of gp130 conditional knockout mice after transaortic constriction (TAC). Control mice (CNT) mice were subjected to a sham operation (squares) or transaortic constriction (diamonds). Mice with conditional ventricular restricted knockout of the gp130 signaling pathway (CKO) were subjected to a sham operation (triangles) or transaortic constriction (circles). Differences in survival rates between the CNT and gp130 conditional knockout mice after TAC were significant by the Peto-Peto-Wilcoxon test (* = p <0.001). (B) Pathological analysis of the gp130 conditional knockout mice hearts. Histological sections of hearts from 3 days and 7 days after transaortic constriction were found in wild-type and gp130 conditional knockout mice. Histological examination of the hearts demonstrated ventriclar enlargement involving both the right and left ventricular chambers in the gp130 conditional knockout mice. (C) A DNA laddering assay (TUNEL) revealed evidence of increased apoptosis in the gp130 conditional knockout mice after (D) low-power (100×) and (E) high-power (1,000×) images showing DNA labeling visualized by fluoresence (green) and counterstained with Hoechst dye (blue). In agreement with the DNA laddering assay, there was significant increase in apoptotic cells in the hearts from the gp130 conditional knockout mice after transaortic constriction, with apoptotic indices of 34% ± 11% in gp130 conditional knockout mice after transaortic constriction versus 3% ± 0.5% in control mice following transaortic constriction (p <0.05). (Modified from Hirota H, Chen J, Betz UA, et al. Loss of a gp130 cardiac muscle cell survival pathway is a critical event in the onset of heart failure during biomechanical stress. Cell. 1999;97:189–198 , with permission.) |
Maladaptive effects of Cytokine Signaling in the Heart
The interest in understanding the role of inflammatory mediators in a variety of cardiac disease states, including heart failure, arises from the observation that many aspects of the syndrome of heart failure can be explained by the known biological effects of proinflammatory cytokines (Table 13-2). When expressed at sufficiently high concentrations,
such as those that are observed in heart failure, cytokines are sufficient to mimic some aspects of the so-called heart failure phenotype, including (but not limited to) progressive left ventricular (LV) dysfunction, pulmonary edema, LV remodeling, fetal gene expression, and cardiomyopathy (29,30,31). Thus, the cytokine hypothesis (32) for heart failure holds that heart failure progresses, at least in part, as a result of the toxic effects exerted by endogenous cytokine cascades on the heart and the peripheral circulation. It should be emphasized that the cytokine hypothesis does not imply that cytokines cause heart failure per se but, rather, that the overexpression of cytokine cascades contributes to disease progression of heart failure. Thus, the elaboration of cytokines, much like the elaboration of neurohormones, may represent a biological mechanism that is responsible for worsening heart failure. Although the deleterious effects of cytokines on myocardial function have received the most attention thus far, cytokines may also produce deleterious effects on LV structure (remodeling) and endothelial function. Accordingly, in the following section, the studies that form the scientific basis for studying the role of proinflammatory mediators in the failing heart will be discussed.
such as those that are observed in heart failure, cytokines are sufficient to mimic some aspects of the so-called heart failure phenotype, including (but not limited to) progressive left ventricular (LV) dysfunction, pulmonary edema, LV remodeling, fetal gene expression, and cardiomyopathy (29,30,31). Thus, the cytokine hypothesis (32) for heart failure holds that heart failure progresses, at least in part, as a result of the toxic effects exerted by endogenous cytokine cascades on the heart and the peripheral circulation. It should be emphasized that the cytokine hypothesis does not imply that cytokines cause heart failure per se but, rather, that the overexpression of cytokine cascades contributes to disease progression of heart failure. Thus, the elaboration of cytokines, much like the elaboration of neurohormones, may represent a biological mechanism that is responsible for worsening heart failure. Although the deleterious effects of cytokines on myocardial function have received the most attention thus far, cytokines may also produce deleterious effects on LV structure (remodeling) and endothelial function. Accordingly, in the following section, the studies that form the scientific basis for studying the role of proinflammatory mediators in the failing heart will be discussed.
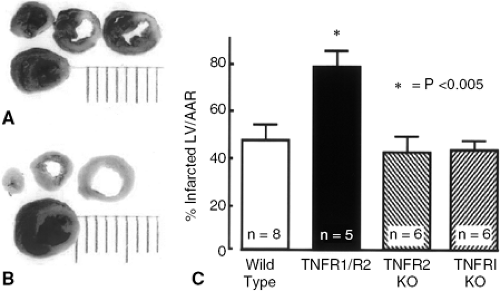 Figure 13-2 Effect of acute coronary artery ligation in TNFR1/TNFR2 knockout mice. The triphenyltetrazolium chloride (TTC) staining deficit, a marker of infarct size, was significantly greater in mice lacking both TNF receptors (B) when compared to littermate control mice (A). The results of group data show that the TTC staining deficit was similar in wild-type mice and mice lacking either the type 1 or type 2 TNF receptors; however, there was a 40% increase in infarct size in the mice lacking both TNF receptors (C). TNFR1/TNFR2 KO, TNFR1/TNFR2 knockout mice; TNFR1 KO, TNFR1 knockout mice; TNFR2 KO, TNFR2 knockout mice). (Modified from Kurrelmeyer K, Michael L, Baumgarten G, et al. Endogenous myocardial tumor necrosis factor protects the adult cardiac myocyte against ischemic-induced apoptosis in a murine model of acute myocardial infarction. Proc Natl Acad Sci USA. 2000;290:5456–5461 .) |
Effects of Cytokines on Left Ventricular Function
The negative inotropic effects of TNF have been observed in studies in which rats were infused with pathophysiologically levels of TNF, as well as in transgenic mice with targeted ovexpression of TNF (29,33,34). Franco et al. used cinemagnetic resonance imaging to demonstrate that there was a significant increase in LV volume and a significant decrease in LV ejection fraction over time in transgenic mice with targeted overexpression of TNF (33). Importantly, these effects were shown to be dependent upon gene dosage. That is, when the line of transgenic mice with high TNF expression (lineage 1) was compared to a transgenic line with lower myocardial TNF expression (lineage 2), there was a significantly greater increase in LV volume (Fig. 13-3A) and a significantly greater decrease in LV ejection fraction (Fig. 13-3B) in the transgenic mouse lines with higher TNF expression (33).
In studies from our laboratory, we measured LV function in line of transgenic mice with targeted overexpression of TNF (34) using Millar catheters. The animals were paced from the atrium to a heart rate at which dP/dt was maximal, as defined by examination of the force-frequency curves for each animal, and peak positive and negative dP/dt were assessed for TNF transgenic mice and littermate controls. As shown in Figure 13-3C, there was significant decrease in peak +dP/dt and peak –dP/dt in the TNF transgenic mice, consistent with the findings reported by Franco et al. (33). Experimental studies in rats have shown that circulating concentrations of TNF that overlap those observed in patients with heart failure are sufficient to produce persistent negative inotropic effects that are detectable at the level of the cardiac myocyte; moreover, the negative inotropic effects of TNF were completely reversible when the TNF infusion was stopped (29). Subsequent studies in transgenic mice with targeted overexpression of TNF in the cardiac compartment have shown that forced overexpression of TNF results in depressed LV ejection performance and that the depressed LV ejection performance was dependent on TNF gene dosage (31,33).
With respect to the potential mechanisms for the deleterious effects of TNF on LV function, the literature suggests that TNF modulates myocardial function through at least two different pathways: an immediate pathway that is manifest within minutes and is mediated by activation of the neutral sphingomyelinase pathway (35) and a delayed pathway that requires hours to days to develop and is mediated by nitric oxide (36,37). Recently, it has been suggested that TNF and IL-1 may produce negative inotropic effects indirectly through activation and/or release of IL-18, which is a recently described member of the IL-1 family of cytokines (38). Relevant to the present discussion is the observation that specific blockade of IL-18 using neutralizing IL-18 binding protein leads to an improvement in myocardial contractility in atrial tissue that was subjected ischemia reperfusion injury (39). Although the signaling pathways that are responsible for the IL-18-induced negative inotropic effects have not been delineated thus far, it is likely that they will overlap those for IL-1, given that the IL-18 receptor complex utilizes components of the IL-1 signaling chain, including IL-IR-activating kinase (IRAK) and TNFR-associated factor-6 (TRAF-6) (38). IL-6 has been shown to decrease cardiac contractility via a nitric oxide (NO)-dependent pathway that is secondary to IL-6-induced phosphorylation of signal transducer and activator of transcription 3 (STAT3). In this study, IL-6 enhanced
de novo synthesis of inducible nitric-oxide synthase (iNOS) protein, increased NO production, and decreased rat cardiac myocyte contractility after 2 hours of incubation. The effects of IL-6 on iNOS production and myocyte contractility were blocked by genistein at concentrations that were sufficient to block IL-6-induced activation of STAT3. Taken together, these observations suggest that IL-6 is sufficient to produce negative inotropic effects through STAT3-mediated activation of iNOS (40).
de novo synthesis of inducible nitric-oxide synthase (iNOS) protein, increased NO production, and decreased rat cardiac myocyte contractility after 2 hours of incubation. The effects of IL-6 on iNOS production and myocyte contractility were blocked by genistein at concentrations that were sufficient to block IL-6-induced activation of STAT3. Taken together, these observations suggest that IL-6 is sufficient to produce negative inotropic effects through STAT3-mediated activation of iNOS (40).
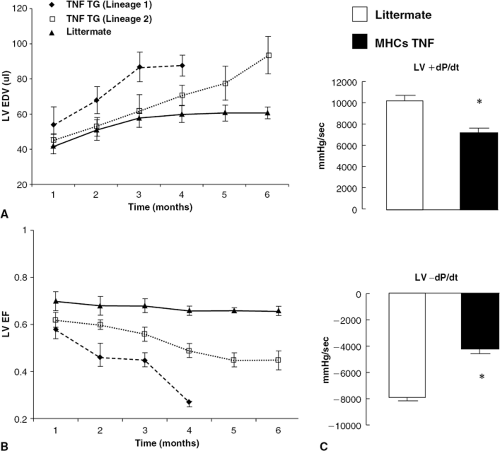 Figure 13-3 Effect of TNF on left ventricular function. LV volume and LV ejection fraction were serially examined by magnetic resonance imaging in two lines of transgenic mice (TNF TG) with high (lineage 1) and low (lineage 2) levels of myocardial TNF expression in comparison to age-matched, littermate control mice (33). (A) Serial changes in LV volume in transgenic mice and littermate control mice. (B) Serial changes in LV ejection fraction in transgenic mice and littemate control mice. (C) LV contractility in mice with targeted overexpression of TNF (MHCsTNF). (From Sivasubramanian N, Coker ML, Kurrelmeyer K, et al. Left ventricular remodeling in transgenic mice with cardiac restricted overexpression of tumor necrosis factor. Circulation. 2001;2001:826–831.) For these studies the animals were paced via the atrium to a heart rate at which positive dP/dt was maximal, as defined by examination of the force-frequency curves for each animal, and peak positive and negative dP/dt were assessed for MHCs TNF mice and littermate controls. (A and B were reproduced from Franco F, Thomas GD, Giroir BP, et al. Magnetic resonance imaging and invasive evaluation of development of heart failure in transgenic mice with myocardial expression of tumor necrosis factoralpha. Circulation. 1999;99:448–454 , and the American Heart Association, with permission.) |
Effects of Proinflammatory Cytokines on Left Ventricular Remodeling
The term left ventricular remodeling has been used to describe the multitude of changes that occur in cardiac shape, size, and composition in response to myocardial injury. As shown in Table 13-3, inflammatory mediators have a number of important effects that may play an important role in the process of LV remodeling, including myocyte hypertroephy
(28), alterations in fetal gene expression (30,31), as well as progressive myocyte loss through apoptosis (41).
(28), alterations in fetal gene expression (30,31), as well as progressive myocyte loss through apoptosis (41).
Table 13-3 Effects of Inflammatory Mediators on Left Ventricular Remodeling | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
In addition to these effects, there are several lines of evidence that suggest that TNF may promote LV remodeling through alterations in the extracellular matrix. First, when concentrations of TNF that overlap those observed in patients with heart failure are infused continuously in rats, there is a time-dependent change in LV dimension that is accompanied by progressive degradation of the extracelluar matrix. Moreover, similar findings have been reported following a single infusion of TNF in dogs (42). Second, recent studies in transgenic mice with targeted overexpression of TNF have shown that these mice develop progressive LV dilation. For example, Kubota et al. showed that a transgenic mouse line that overexpressed TNF in the cardiac compartment developed progressive LV dilatation over a 24-week period of observation (31). Similar findings have also been reported by Bryant et al. (43) and Sivasubramanian et al. (34), who observed identical findings with respect to LV dysfunction and LV dilation in transgenic mice with targeted overexpression of TNF in the heart. With respect to the mechanisms that are involved in TNF-induced LV dilation, it has been suggested that TNF-induced activation of matrix metalloproteinases (MMPs) is responsible for this effect (34,44).
As shown in Figures 13-4 and 13-5, respectively, there was progressive loss of fibrillar collagen and increased
MMP activation in the hearts of the transgenic mice overex-pressing TNF in the cardiac compartment. The dissolution of the fibrillar collagen weave that surrounds the individual cardiac myocytes and links the myocytes together would be expected to allow for rearrangement (slippage) of myofibrillar bundles within the ventricular wall (45). However, Figure 13-5 shows that long-term stimulation (i.e., 8 to 12 weeks) with TNF resulted in an increase in fibrillar collagen content that was accompanied by decreased MMP activity (Fig. 13-5) and increased expression of the tissue inhibitors of matrix metal toprotemases (TIMPs) (Fig. 13-5).
MMP activation in the hearts of the transgenic mice overex-pressing TNF in the cardiac compartment. The dissolution of the fibrillar collagen weave that surrounds the individual cardiac myocytes and links the myocytes together would be expected to allow for rearrangement (slippage) of myofibrillar bundles within the ventricular wall (45). However, Figure 13-5 shows that long-term stimulation (i.e., 8 to 12 weeks) with TNF resulted in an increase in fibrillar collagen content that was accompanied by decreased MMP activity (Fig. 13-5) and increased expression of the tissue inhibitors of matrix metal toprotemases (TIMPs) (Fig. 13-5).
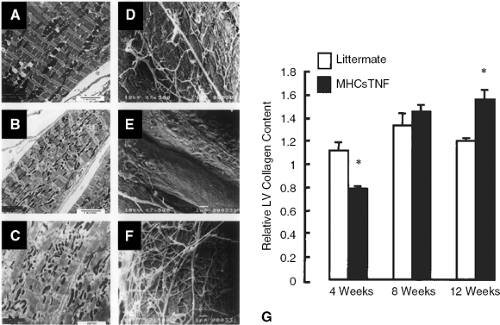 Figure 13-4 Effects of sustained proinflammatory cytokine expression on myocardial ultrastructure and collagen content. A-C show representative transmission electron micrographs in littermate controls (A) and the TNF transgenic mice at 4 weeks (B) and 8 weeks of age (C). The transmission electron micrographs from the littermate control mice at 4 weeks (A) revealed a characteristic linear array of sarcomere and myofibril. In contrast, the myofibril in the 4-week-old TNF transgenic mice were less organized, with loss of sarcomeric registration observed in many of the sections (B). The ultrastructural abnormalities in the TNF transgenic mice were further exaggerated in the 12-week-old TNF transgenic mice, which showed a significant loss of sarcomere registration and myofibril disarray (C). D-F show representative scanning electron micrographs in littermate controls (D) and the TNF transgenic mice at 4 weeks (E) and 8 weeks of age (F). E shows that there was a significant loss of fibrillar collagen in the TNF transgenic mice at 4 weeks of age when compared to age-matched, littermate controls (D). However, as the TNF transgenic mice aged (12 weeks), there was an obvious increase in myocardial fibrillar collagen content. G illustrates the myocardial collagen content as determined by picrosirius red staining. There was a loss of myocardial collagen content at 4 weeks of age in the TNF transgenic mice, which was later followed by a progressive increase in myocardial collagen content at 8 and 12 weeks of age. (Reproduced from
Get Clinical Tree app for offline access
Sivasubramanian N, Coker ML, Kurrelmeyer K, et al. Left ventricular remodeling in transgenic mice with cardiac restricted over-expression of tumor necrosis factor. Circulation. 2001;2001: 826–831
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree


|