Fig. 10.1
Age distribution of deaths from cystic fibrosis in the UK 2009. Nowadays very few deaths occur in childhood, the majority occur in early adulthood, and an increasing number occur in middle to older age (Reproduced with permission from the Cystic Fibrosis Trust annual data report [1])
Overview of Cystic Fibrosis
CF is an autosomal recessive disease caused by mutations of a gene on chromosome 7 that encodes for a protein named cystic fibrosis transmembrane conductance regulator (CFTR), which functions as a chloride channel in the apical membrane of epithelial cells [4]. Reduced chloride conductance results in viscid secretions and organ damage in the respiratory, gastrointestinal, hepatobiliary, and reproductive tracts. It affects about 1 in 2,500 births in the UK, and about 1 in 25 of the population is a carrier of the disease.
More than 1,800 mutations of the CF gene have been identified and this partly explains the wide spectrum of severity of the disease, although other factors such as environmental factors and modifier genes affecting the inflammatory response are also important. The most common mutation, traditionally known as ΔF508 (F508del), results in the loss of phenylalanine at position 508 of the protein. This causes misfolding of the mutant CFTR, which is then degraded such that no CFTR reaches the cell membrane. In certain mutations, the mutant CFTR retains some function and this may be associated with less severe disease. Some rare patients have non-classic CF in which they have two mutations of the gene, abnormal sweat chloride, some manifestations of the disease (such as male infertility, pancreatitis, and nasal polyps) but little or no lung disease. There is a spectrum of severity and the treatment regimen is adjusted according to the type, severity, stage, and complications of the disease.
The CF gene has been cloned and given to patients using liposomes or inactivated viruses as gene transfer agents. Expression of the gene has been confirmed by measuring transepithelial potential difference and trials are in progress to assess whether gene therapy can improve clinical outcomes. Substantial progress is being made in treatments that address the molecular biology processes of CFTR transcription from DNA, trafficking through the cell and activation and regulation at the cell membrane [4]. Some treatments are targeted at specific mutations. Thus a small molecule, named Ataluren®(PTC Therapeutics), induces ribosomes to read through premature termination codons during mRNA translation of certain mutations (such as G542X and W1282X). Ivacaftor®(Vx770; Vertex Pharmaceuticals) is a CFTR potentiator that improves CFTR function in patients with the G551D mutation. These new treatments are likely to improve the outcome for patients now being born with CF, but are likely to be of less benefit to those who already have advanced lung disease.
In the bronchial mucosa, reduced chloride secretion and increased sodium re-absorption result in secretions of abnormal viscosity with reduced water content of the airway surface liquid and disrupted mucociliary clearance. This predisposes to chronic infection with associated inflammation progressing to bronchiectasis, respiratory failure, and death. Initially infection is often withStaphylococcus aureus. Subsequently,Pseudomonas aeruginosabecomes the dominant pathogen. Different strains of pseudomonas can be identified and they vary in their virulence and transmissibility.Burkholderia cepaciacomplex is a group of Gram-negative bacteria which have a high level of antibiotic resistance. Patients with CF are vulnerable to these bacteria and infection can spread from patient to patient. The most virulent strains areB cenocepaciaandB multivorans. Non-tuberculous mycobacteria (such asMycobacterium abscessus) can also infect the lungs in CF and are difficult to treat. Patients are segregated according to their infections when attending hospital and contact between patients is discouraged.
In the pancreas, plugging and obstruction of ductules causes progressive destruction of the gland. Pancreatic enzymes (such as lipase) fail to reach the small intestine and this results in malabsorption of fats with steatorrhea and weight loss. Approximately 40 % of adults develop diabetes from destruction of the endocrine pancreas. Lack of pancreatic enzymes with viscid intestinal secretions and reduced motility can result in distal intestinal obstruction, which is usually treated by intestinal lavage and osmotic agents (such as Gastrografin ®). Men with CF are nearly always infertile because of obstruction of the vas deferens but can achieve biological parenthood by sperm aspiration from the testes with in vitro fertilization. Women with CF have essentially normal fertility and many undertake pregnancy and motherhood, but may not survive to see the child reach adulthood.
The key elements of treatment are clearance of bronchial secretions by physiotherapy, treatment of pulmonary infection by antibiotics, and correction of nutritional defects by pancreatic enzyme supplements and dietary support [5]. Patients are typically given flucloxacillin continuously to prevent or suppressStaphylococcus aureusinfection. When Pseudomonas is first isolated, attempts are made to eradicate it by prolonged courses of oral ciprofloxacin and nebulized colistin or tobramycin. When pseudomonas infection becomes established, long-term nebulized antibiotics are used to suppress infection. Exacerbations are treated by combinations of intravenous antibiotics (such as ceftazidime or meropenem with tobramycin or colistin). Treatment is facilitated by use of indwelling central venous access devices and many patients self-administer antibiotics intravenously at home (Fig.10.2). As the disease progresses, the frequency of antibiotics increases. Even in late stage disease intravenous antibiotics are effective in improving the level of symptoms. Malabsorption is controlled by use of pancreatic enzymes with food. As the chest disease progresses, patients have difficulty in maintaining their energy requirements because of decreased appetite and the increased energy expenditure associated with chronic lung infection. Dietary supplements are used when anorexia limits intake and supplemental feeding is often given overnight via a gastrostomy tube. Mucolytic agents, such as nebulized 7 % saline or deoxyribonuclease (DNase), aid clearance of airway secretions. Lung transplantation is the main treatment for patients with end-stage lung disease but lack of donor organs limits this treatment. Overall survival rates post-transplantation are approximately 80 % at 1 year, 60 % at 5 years, and 50 % at 10 years [6]. Palliative care is paradoxically intertwined with high-intensity treatments including lung transplantation and many patients who die will be on a transplant waiting list at the time of death.
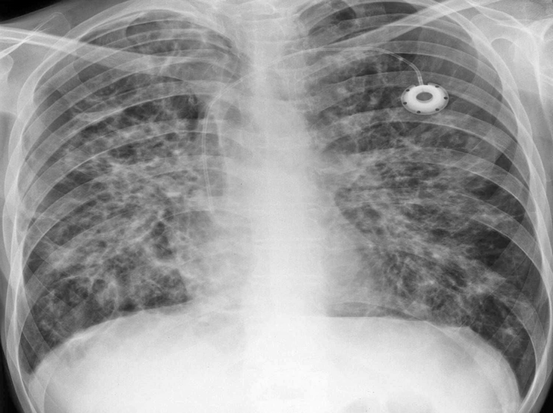
Fig. 10.2
This man died at 21 years of age of respiratory failure from progressive cystic fibrosis lung disease. Two years previously he had been deemed unsuitable for lung transplantation because ofBurkholderia cenocepaciainfection. He had a prolonged phase of palliative care of advanced disease during which he had supplemental gastrostomy feeding, intensive physiotherapy, and frequent courses of intravenous antibiotics. He also attended a specialist palliative care symptom-control clinic, but he did not find a hospice day care facility helpful. His chest radiograph shows severe diffuse bronchiectasis and a central venous access system in the left subclavian vein. After several exacerbations he deteriorated into terminal stage disease and died at home with end of life care from the community palliative care team
Predicting the Prognosis
Patients with CF live their lives in the knowledge of having a chronic life-limiting disease. They show remarkable resilience in the face of the disease: “I do not sit around worrying about when I am going to die. I will worry about the future when it arrives. I think about it but do not walk around clutching my funeral arrangements” [7]. At present the median survival is about 38 years but it is predicted to be at least 50 years for children now being born with the disease [8]. In most patients, there is a gradual, generally predictable progression of lung disease that allows a planned approach to both lung transplantation and palliative care. However, patients can suffer an acute crisis from complications such as massive hemoptysis, pneumothorax, or a severe acute exacerbation. Lung transplantation is generally considered at a stage where the forced expiratory volume in one second (FEV1) has fallen to below 30 % of the predicted value. Some studies suggest that this indicates an approximately 50 % risk of dying within 2 years [9]. Additional factors such as the rate of decline, frequency of exacerbations, development of hypoxia and hypercapnia, and occurrence of complications are also important in predicting a poor prognosis. However, the advanced stage of the disease is often a prolonged phase over many years, and intensive treatment with frequent courses of intravenous antibiotics, mucolytic agents, physiotherapy, and nutritional support can improve the survival and quality of life for these patients [10]. Some patients are unsuitable for transplantation because of resistant infections (such asBurkholderia cenocepacia,Mycobacterium abscessus), poor nutritional status, or additional systemic disease (such as renal disease). Some patients do not wish to undergo transplantation because they fear high-risk intensive treatments. The shortage of donor organs means that up to 40 % of patients considered suitable for transplantation die before donor lungs become available [11].
As with many chronic lung diseases, there is often a prolonged phase of advanced disease requiring symptom control and palliative supportive care followed by a short terminal phase. Precise prediction of the likely time of death is often not possible or helpful, and many of these patients have already survived beyond the life expectancy given to them in childhood. Often it is better to acknowledge that when the disease is progressing and transplantation is not feasible the aim of treatment is to keep patients “as well as possible for as long as possible and to focus on keeping them comfortable when the time comes.” This equates to a goal of optimizing the quality of life and ensuring a peaceful, dignified natural death. Many will have specific aims which they want to achieve within their limited life expectancy, such as bringing-up a young child. The classic disease trajectory of cancer patients, with a gradual switch from active treatments to palliation over a relatively short time period, does not apply to patients with CF where palliative care principles are applicable at all stages of the disease, run in parallel with disease-modifying treatments and are still appropriate for patients who ultimately undergo a rescue lung transplant. In a cancer trajectory, care will often transfer from the medical team to the palliative care team, whereas in the chronic illness trajectory integration of palliative care into the multidisciplinary management of the disease is often more appropriate.
Symptom Control in Advanced Disease
In advanced CF, patients often have a range of complex problems. Symptoms can be both physical and psychological. The dominant physical symptoms are cough, sputum, wheeze, chest tightness, breathlessness, pain, fever, and fatigue [12,13]. The emotional impact of these symptoms is high, and when combined with a realization that the prognosis is poor, results in frustration, anger, sadness, irritability, worry, and difficulty sleeping. A study of self-reported symptom burden revealed that patients had a median of ten different symptoms [13]. These symptoms and the emotional response impact on activities with more time spent sitting or lying, a reduction in usual activities, and missing work. Pain is a common symptom at all stages of CF but becomes more prominent as the disease progresses. Surveys show that 84 % of patients with advanced CF have pain: 65 % have chest pain, 55 % headaches, 19 % back pain, and 19 % abdominal pain [14]. The majority of chest and back pain is of musculoskeletal origin related to use of accessory muscles of respiration, coughing, postural abnormalities, or osteoporosis. Several studies suggest that the problems of chronic pain in CF are not adequately addressed with a lack of use of analgesic medication or other treatment strategies for pain [14,15]. When assessing patients with CF in addition to focusing on key elements of the disease such as lung function, oxygenation, nutritional status, and control of infection, it may also be helpful to use symptom check lists to identify the symptom burden, the emotional impact of these symptoms, and the impact these symptoms are having on activities and quality of life [12,13]. Symptom control requires detailed assessment and a multifaceted approach. Specific treatments directed against the disease, such as a course of intravenous antibiotics, result in improvements in quality of life and in multiple domains of the chronic disease questionnaire [12]. Some symptoms, such as hemoptysis, may require a specific intervention such as bronchial artery embolization. Other specific interventions targeting specific symptoms include analgesia for pain, sputum clearance physiotherapy for cough and secretions, mucolytics for sputum retention, and anti-emetics for nausea and vomiting. Management of psychological symptoms should be part of a holistic approach.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


