Figure 3.1
Survival plot before and after the advent of targeted vasodilator therapy, using REVEAL registry data (2012) of idiopathic and familial PAH matched to that from National Institutes of Health registry (1980s) (Adapted from Benza et al. Chest [6] with permission)
Endothelin Receptor Antagonists
Endothelin-1 is a peptide produced by the endothelium with potent vasoconstricting and smooth muscle proliferating properties. It is found at higher concentrations in pulmonary hypertension and has also been implicated in the pathogenesis of atherosclerosis and systemic hypertension. Oral endothelin receptor antagonism has been used since the late 1990s in pulmonary arterial hypertension.
The two receptor isoforms A and B are more prominent in pulmonary artery smooth muscle and vascular endothelium respectively. Bosentan and macitentan act on both receptors whereas ambrisentan (and sitaxentan) are selective for endothelin receptor A. Despite endothelin receptor B having vasodilatory effects and playing a role in endothelin-1 clearance, the selective antagonists have not been shown to be clinically superior to date.
Transaminitis is the most important side-effect and hence monthly blood test monitoring is required. Sitaxentan was initially reported to be efficacious and to have a low incidence of hepatic complications [7] but was taken off the market in 2010 due to several reported deaths from hepatotoxicity [8].
Other side-effects of these agents are peripheral oedema, nausea, hypotension, teratogenicity, anaemia, headaches/jaw pain and flushing. Ambrisentan needs to be used with caution in idiopathic pulmonary fibrosis [9].
Bosentan
The BREATHE-1 trial was a 2002 randomised controlled trial (RCT) with 213 idiopathic and associated PAH patients who were highly symptomatic, namely functional class (FC) III or IV. After 16 weeks’ treatment, the mean 6 min walk test (6MWT) increased by 35 m and 54 m (125 mg and 250 mg BD respectively versus placebo), and 34–38 % had improved to WHO FC II. In addition the time to clinical worsening was prolonged. There was a 7 % reported transaminitis rate at the higher dose [10].
The EARLY study from Italy in 2008 recruited 185 FC II, group 1 patients. After 6 months, the haemodynamics and symptoms improved and time to clinical worsening was delayed. Pulmonary vascular resistance (PVR) was 83 % of baseline in the treated arm and 6MWT 19 m further between the two arms [11].
Ambrisentan
ARIES 1 and 2 were concurrent double-blind trials involving a total of 394 idiopathic and associated PAH patients who were targeted vasodilator treatment naive and had a 6MWT between 150 and 450 m. The randomisation was to different dosing regimens (2.5, 5 and 10 mg OD) and placebo controlled. The primary end-point of 6MWT increased by between 31 and 54 m with dose dependency seen. Also seen was statistically significant improvement in functional class, health related questionnaire, time to clinical worsening, Borg dyspnoea and BNP level. The initial study time period was 12 weeks but an extension studies showed persistent benefits to 48 weeks and 2 years. Elevation in transaminases was quoted at 3.9 %. Hence ambrisentan is considered to have a safer liver side-effect profile than bosentan [12, 13].
ARIES 3 was an open label, uncontrolled study that broadened the patient selection criteria to include Dana Point groups 3 and 4. Overall 6MWT increased by 21 m. This improvement was seen in groups 1 and 4 but not group 3. The COPD and ILD sub-group demonstrated a slight deterioration in their exercise capacity but their BNP levels did decrease in keeping with the other aetiologies [14].
Macitentan
This novel unselective endothelin receptor antagonist is believed to be more potent due to prolonged binding time and tissue penetration. Macitentan had initially shown promising results in animal models and now has a large international RCT with morbidity benefit supporting its use.
The SERAPHIN study of 742 group 1 patients, with the primary outcome measure of morbidity events and mortality, was reported in 2013 [15]. The study population were mainly FC II and III and approximately 60 % were already taking targeted vasodilator medication, predominantly a phosphodiesterase-5 inhibitor. There was a highly significant reduction in adverse events, usually clinical worsening of PH, and the hazard ration for the 10 mg dose of macitentan compared with placebo was 0.55. There was also a non-significant trend towards mortality benefit. 6MWT and FC also showed significant improvement (the former by 22 m) with a degree of dose dependency. The transaminitis incidence was lower than placebo at 3.5 %.
Phosphodiesterase-5 Inhibitors
Sildenafil, tadalafil and vardenafil are selective cyclic GMP phosphodiesterase type 5 inhibitors which are administered orally. cGMP acts as a second messenger in the nitric oxide pathway and PDE5 is upregulated in the pulmonary hypertension vascular bed. The PDE5 inhibitors prevent the breakdown of cGMP and hence cause potentiation of the endothelial smooth muscle relaxtion of NO. In addition they are believed to be anti-proliferative and have shown pulmonary vascular remodelling in vitro.
Side-effects include postural hypotension (hence the contraindication with concurrent nitrates), visual disturbance including cyanopsia (with cGMP PDE5 particularly prevalent in the retina), headache, hearing loss, flushing and dyspepsia. They are the most recently developed class, with sildenafil first licensed in 2005. Two newer PDE5 inhibitors, tadalafil and vardenafil, are longer acting (hence requiring less frequent administration) and appear to be equally efficacious. Vardenafil may be more potent due to prolonged binding time.
Sildenafil
Case reports, small non-randomised studies and one RCT from the early 2000s first showed the benefit of sildenafil [16–20] but the strongest evidence for its use as monotherapy comes from the SUPER-1 trial. 278 PAH patients were recruited into a 12 week double blind RCT with four arms, placebo versus 20, 40 and 80 mg TDS. The population studied were IPAH, connective tissue disease and a few congential patients post-surgical correction. There was improvement in 6MWT (improved by 45–50 m, which represented 13.0–14.7 %), mean PA pressure (small absolute reduction but statistically significant) and WHO FC (improved by at least one in 7 % of placebo and up to 42 % at the highest dosage) [21]. The same trial population also undertook health related quality of life questionnaires SF-36 and EQ-5D with significant improvement [22].
259 of the above patients underwent a 3 year open label, uncontrolled extension in SUPER-2. Approximately half at least maintained their 6MWT and functional class over the 3 years from pre-treatment baseline. 3 years survival was 79 %. The majority were uptitrated to 80 mg TDS (with good safety profile) and 18 % had had a second agent added by the end [23].
Tadalafil
The PHIRST RCT involved 405 idiopathic or associated PAH patients. The highest dose 40 mg OD increased 6MWT by a mean 44 m after 16 weeks as single agent [24].
Vardenafil
Vardenafil was studied by RCT in treatment naive PAH over 12 weeks in the EVALUATION trial. 6MWT improved by 69 m. Haemodynamics were also significantly improved in terms of pulmonary artery pressure (down 5.3 mmHg), cardiac output (up by 0.39 l/min) and pulmonary vascular resistance (down by 4.7 WU). WHO FC and Borg dyspnoea were also improved. For the first time a PDE5 inhibitor was shown to reduce clinical worsening events, namely death or hospitalisation [25].
Prostacyclin Analogues
Prostacyclin (or prostaglandin I2) is a prostinoid produced by vascular endothelium that has vasodilatory, anti-thrombotic, anti-proliferative and anti-inflammatory properties. The pathway has a significant role in the pathogenesis of pulmonary hypertension. Intravenous epoprostenol was the first advance targeted vasodilator licensed for treatment of PH (in 1998). Several analogues have been developed but the mode of administration remains problematic.
Epoprostenol
Initial trials from the 1990s showed benefit from continuous IV epoprostenol infusion in idiopathic pulmonary arterial hypertension [26–29].
As with the other classes, the best level of evidence comes from group 1 PAH patients who have advanced disease, i.e. WHO FC III or IV. The above unblinded RCT by Barst et al. in 1996 involved 81 such patients and showed statistically significant improvement in terms of exercise tolerance (6MWT differential of 47 m between the arms and functional class improved in 40 % compared with only 3 % in the placebo arm), haemodynamics (PVR reduced by 21 % compared with a 9 % increase in placebo) and even a survival benefit (zero deaths compared with 8) over a 12 week period.
Epoprostenol has a half-life of only a few minutes and consequently does require continuous infusion without interuption. The syringe needs to be changed every 8 h due to the medication degrading unrefrigerated. Specific complications include pump disruption, infusion pain, line thrombosis and infection. Should there be any disruption to the flow then there is a 10–15 min window to rectify matters. If this is unsuccessful then there is the potential for a rebound pulmonary hypertensive crisis and death. Other complications include hypotension, jaw pain, flushing, wheeze, nausea, diarrhoea, agitation and arthralgias.
Treprostinil
Treprostinil is an epoprostenol analogue which has been developed to have greater stability at room temperature. Consequently it has the advantage that is can be given subcutaneously as well as intravenously. Subcutaneously it has been shown to have a survival benefit and improved functional class in PAH patients [30]. Oral preparations are being developed but their efficacy has not been proven to date [31].
Iloprost
Inhaled or nebulised iloprost has been shown to be beneficial in a range of aetiologies including group 1 and group 4 with FC III and IV. It has to be administered every 2–4 h and proves a significant time burden on those using it [32]. There is only limited evidence with the use of intravenous iloprost with its efficacy called into question.
Beraprost
Selexipag
This novel prostacyclin receptor agonist can be administered orally and has phase 2 evidence behind it. It is discussed later in the novel/future therapies section (Table 3.1).
Table 3.1
Summary of advanced, targeted pulmonary vasodilator medication
Endothelin receptor antagonists | ||||||
Side–effects | Hepatic dysfunction, peripheral oedema, nausea, hypotension, teratogenicity, anaemia, headaches/jaw pain and flushing | |||||
Name | Brand name | Route of administration | Initial Dose | Up–titration dose | Monitoring | Notes |
Bosentan | Tracleer | Oral | 62.5 mg BD | 250 mg BD | Monthly liver function required | Interaction with warfarin (bosentan) Contra-indicated with cyclosporin |
Ambrisentan | Volibris | Oral | 5 mg OD | 10 mg OD | ||
Macitentan | Opsumit | Oral | 3 mg OD | 10 mg OD | ||
Phosphodiesterase–5 inhibitors | ||||||
Side–effects | Postural hypotension, visual disturbance including cyanopsia, headache, hearing loss, flushing and dyspepsia | |||||
Name | Brand name | Route of administration | Initial Dose | Up–titration dose | Monitoring | Notes |
Sildenafil | Revatio, Viagra | Oral | 20 mg TDS or 25 mg TDS | 50 mg TDS | History of visual impairment | Contra-indicated with nitrates First line in hepatic dysfunction |
Tadalafil | Adcirca | Oral | 20 mg OD | 40 mg OD | ||
Vardenafil | Levitra | Oral | 5 mg BD | 20 mg BD | ||
Prostacyclin analogues | ||||||
Side–effects | Hypotension, jaw pain, flushing, wheeze, nausea, diarrhoea, agitation and arthralgias. Administration may be complicated by pump disruption, infusion pain, line thrombosis and infection | |||||
Name | Brand name | Route of administration | Initial Dose | Up–titration dose | Monitoring | Notes |
Epoprostenol | Veletri, Flolan | Intravenous | 2 ng/kg/min | 200 ng/kg/min | High risk of rebound pulmonary hypertension if infusion is disrupted | |
Treprostinil | Remodulin, Tyvaso | Intravenous, subcutaneous or nebulised | 1.25 ng/kg/min IV/SC | 40 ng/kg/min IV/SC | ||
Iloprost | Ventavis | Nebulised | 2.5 μg 6–9× per day | 5 μg 6–9× per day | ||
Combination Therapy
In the future it seems likely that dual or even triple therapy will be commenced at an earlier stage. Randomised controlled trials are being reported every year but have had mixed results to date. All of the following studies are exclusively in group 1 PAH patients.
One relatively early prospective cohort showed the benefit of sildenafil added into iloprost in the context of clinical deterioration [35].
Bosentan and prostanoids in combination in idiopathic PAH has shown contradictory results with the following studies being positive and negative respectively [36, 37]. However the best calibre of evidence comes from the 2006 STEP-1 trial with 67 PAH patients randomised to placebo versus iloprost added into bosentan monotherapy. The 6MWT improvement at 12 weeks was 26 m and 34 % improved functional class (compared with 6 % in placebo arm) [38].
BREATHE 2 saw bosentan added into epoprostenol and although there was a trend towards functional improvement with both therapies, it was not significant [39].
Sildenafil plus epoprotenol in 267 class III patients showed significant improvement in a range of clinical and haemodynamic parameters [40].
The PHIRST trial increased 6MWT by 23 m when tadalafil was added into bosentan [24].
The positive SERAPHIN study into macitentan had almost two thirds of its participants already taking a phosphodiesterase-5 inhibitor and revealed a significant morbidity benefit [15].
TRIMUPH in 2010 involved 235 severe patients (functional class III and IV) where inhaled treprostinil was added into bosentan or sildenafil. There was an improvement in the primary end-point of 6 min walk testing (improved by 20 m) but no change in time to clinical worsening or level of dyspnoea [41].
The FREEDOM-C and FREEDOM-C2 trials have been published in the last 12 months. Oral treporostinol, at different dosages, was added to patients already on advanced vasodilators, be that a PDE-5 inhibitor, endothelin receptor antagonist or both. There was no improvement in the primary end-point of 6MWT and there was a 22 % discontinuation rate with high incidence of side-effects. There was however some benefit in secondary outcomes such as dyspnoea [31, 42].
The AMBITION study is evaluating combination ambrisentan and tadalafil versus single agent arms in 614 treatment naive PAH patients and should be publishing in the near future.
Initial triple therapy was evaluated in 10 idiopathic and heritable PAH patients with FC III and IV and haemodynamic severity (cardiac index less than 2 l/min/m2 or pulmonary vascular resistance more than 12.5 Wood units) at diagnosis. The results were very impressive with all patients at 4 months (n of 7) improving to FC II but one requiring transplantation due to lack of improvement. There was a mean improvement in 6MWT of 164 m, mean PA pressure reduction of 13 mmHg, PVR reduction of 16.7 Wood units and cardiac index improvement of 2.l/min/m2 which was maintained at (median) 18 months and well tolerated [43].
Calcium Channel Blockers
Prior to the advent of targeted vasodilators, inhaled nitric oxide response was used as a predictor of response to high dose calcium channel blockers. Those not responding had, in addition to the lack of clinical efficacy, a far higher incidence of side-effects including severe complications such as cardiogenic shock and hypoxaemia (due to loss of hypoxic pulmonary vasoconstriction and subsequent worsening V/Q matching).
Vasoreactivity testing (with inhaled nitric oxide or intravenous adenosine/epoprostenol at the time of right heart catheterisation – RHC) should be performed in all group 1 PH patients. Calcium channel blockers are not used as the testing agent due to the high risk of serious side-effects in the non-responders [44]. Testing is relatively contraindicated in pulmonary veno-occlusive disease due to the risk of pulmonary oedema.
The test is considered positive should the mean PA pressure fall by at least 10 mmHg to a value of under 40 mmHg without any fall in the cardiac output. There is approximately a 10 % chance that this will be the case. This rises significantly in the anorexigen induced PH population. The importance of a positive vasoreactivity test is a significantly better prognosis and it is a strong predictor for good response to calcium channel antagonists. High dose, sustained release preparations of nifedipine and diltiazem (with the strongest evidence base) are most commonly used. Observational studies have shown a symptomatic and survival benefit that can be prolonged over years [3].
There is however the suggestion that responders have less severe disease at baseline with a longer duration of symptoms [45]. Consequently they are likely to represent a distinct, more indolent phenotype and there is evidence to show the survival benefit is seen even when not treated with calcium channel blockers [46].
Close follow-up is suggested to ensure the calcium channel antagonist response is maintained (some guidelines suggesting a repeat RHC at 3–4 months) with only 54 % reported to maintain the beneficial response [45]. Targeted vasodilators should be commenced early when the response is not maintained.
Guidelines
The joint European Society of Cardiology (ESC) and European Respiratory Society (ERS) guidelines were published in 2009 [47]. The World Symposium on Pulmonary Hypertension took place in Nice in February/March 2013. Consequently there have been a number of recently published updates to the classification and treatment regimen [48]. The guidelines are summarised below.
A holistic approach to treatment is suggested including general measures (such as oxygen, diuretics and warfarin, as detailed in general measures), exercise, infection prevention, pregnancy advice, psychosocial support, multi-disciplinary approach and palliative care when appropriate.
There are a number of established treatment goals with an evidence basis behind them. Essentially it orientates around keeping patients in the “stable and satisfactory” group and the prevention or reversal of poor prognostic indicators. The precise therapeutic goals are as follows:
Clinical – no signs of right ventricular failure or history of syncope, WHO functional class I or II, 6MWT >500 m, peak VO2 > 15 ml/min/kg and normal or near-normal BNP;
Echocardiography – no pericardial effusion and TAPSE >2.0 cm;
Right heart catheterisation – right atrial pressure <8 mmHg and cardiac index ≥2.5 l/min/m2.
First line in the treatment algorithm of PAH patients is the vasoreactivity testing and commencement of calcium channel blockers if positive. In non-responders, choice of targeted vasodilator depends on functional class, with treatment usually indicated when in FC II and above. First line choice is not usually specified due to lack of data. Therefore that choice is made by the treating physician with the knowledge of the individual circumstances to guide therapy for example hepatic or renal dysfunction.
However there are some caveats. The one exception to specified therapy is intravenous epoprostenol as first line in FC IV. This is also the only group where combination therapy should be considered from the outset. In FC II an endothelin receptor antagonist or phosphodiesterase-5 inhibitor should be considered first line.
Although not always specified in the guidelines, it would be usual practice to routinely uptitrate the dose of single vasodilator if tolerated. Should there be insufficient clinical improvement then this would trigger the addition of initially a second agent and then full triple therapy. Inadequate response is defined as “stable and not satisfactory” (i.e. therapeutic goals not met) or “unstable and deteriorating” (such as the development of right heart failure) in FC II or III. At the most severe end of the spectrum, there is also emphasis on rapidly leaving FC IV.
The precise treatment algorithm with grades of supporting evidence is reproduced below (Fig. 3.2).
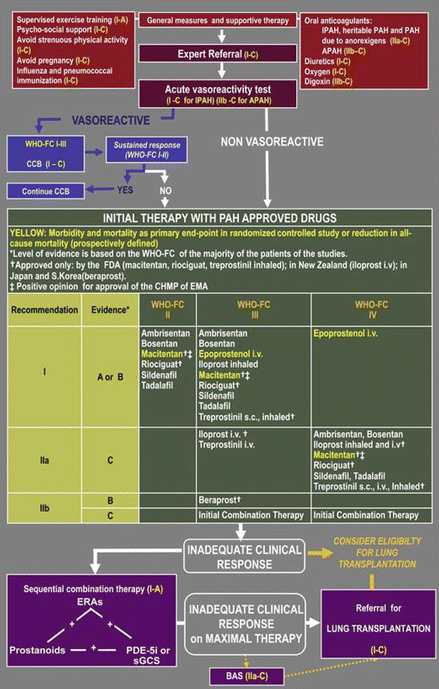
Figure 3.2
Evidence-based treatment algorithm for pulmonary arterial hypertension patients (for group 1 patients only) (Reproduced from Galiè et al., Journal of the American College of Cardiology [48] with permission) Key: APAH associated pulmonary arterial hypertension, BAS balloon atrial septostomy, CCB calcium channel blockers, ERA endothelin receptor antagonist, sGCS soluble guanylate cyclase stimulators, IPAH idiopathic pulmonary arterial hypertension, i.v. intravenous, PDE-5i phosphodiesterase type-5 inhibitor, s.c. subcutaneous, WHO-FC World Health Organization functional class
There are also guidelines from the American College of Cardiology Foundation and American Heart Association published in 2009 [49]. These are similar to the ESC/ERS guidelines except instead of using functional class to guide treatment, patients are divided into lower and high risk. As per FC IV, high risk patients are suggested to commence intravenous prostanoids but either epoprostenol or treprostinil is suggested. The risk stratification is determined by clinical criteria. High risk criteria being FC IV, rapid progression, right ventricular failure, 6MWT <300 m, peak VO2 < 10.4 ml/kg/min, pericardial effusion, significant right ventricular enlargement or dysfunction, right atrial enlargement, RA pressure >20 mmHg, CI <2.0 l/min/m2 and significantly elevated BNP.
General Measures
Anticoagulation
Several observational, predominantly retrospective, studies performed in the 1980s showed an improvement in survival when taking warfarin in idiopathic pulmonary hypertension. Subgroup analysis from the 1992 calcium channel antagonist study revealed a 3 year survival benefit from 31 to 62 % with warfarin in the non-responder population [3], albeit those selected out for treatment by non-uniformity on a perfusion scan. A 2006 literature review found a survival benefit in 5 out of 7 studies involving idiopathic PAH [50].
The rationale behind anticoagulation is prothrombotic tendency (with abnormal clotting cascade, platelet function and fibrinolysis) and altered right sided haemodynamics (sluggish flow in dilated atrium and ventricle). Thrombotic arteriopathy has a strong histopathological association with pulmonary arterial hypertension although the cause/effect relationship is unknown [51].
Current best practice is to commence warfarin, in the absence of contraindication, in certain group 1 (namely idiopathic, hereditable and anorexigen induced) and naturally group 4 Dana Point pulmonary hypertension. It is not usually recommended in connective tissue disease associated PH due to an increased bleeding risk. The optimal target INR or functional class for treatment has not been established.
There is currently no data (even in CTEPH) regarding the use of the novel oral anticoagulant agents (such as the direct thrombin inhibitor dabigatran and the factor Xa inhibitors rivaroxaban and apixaban) and there are also interactions with endothelin receptor antagonists and phosphodiesterase 5 inhibitors.
Digoxin
Digoxin is often prescribed at a low dose more for its inotropic effect than as an antiarrhythmic. Despite being prescribed in up to half of all patients with pulmonary hypertension, the evidence basis is weak. A small RCT from 1981 in cor pulmonale (Group 3) patients did show an improvement in right ventricular function after 8 weeks but only when in the context of biventricular dysfunction [52].
Acute administration of digoxin at time of right heart catheterisation shows physiological improvement in idiopathic PAH. There is haemodynamic improvement, namely a 10 % increase in cardiac output with no change in pulmonary vascular resistance, and also a reduction in circulating levels of noradrenaline [53]. There is however no long term data supporting the use of digoxin in pulmonary hypertension.
Beta Blockers
Beta-blockers have been considered relatively contraindicated in pulmonary arterial hypertension due to the negative inotropic and chronotropic effects impacting on right ventricular function. Due to the inability to increase stroke volume, the PH cardiac output in exercise is predominantly rate dependent [54].
In addition beta-blockers may cause pulmonary vasoconstriction as suggested by animal models. However 12 % of group 1 patients were taking beta-blockers in the REVEAL registry [55].
Contrary to the above, more recent evidence suggests beta-blockade may be safe in PAH. First beta-blockers were found to reverse right ventricular remodelling and improve function and cardiac output in a rat model [56]. A prospective controlled cohort of 94 patients of group 1 PAH patients revealed no statistical difference in clinical end-points including mortality and right sided heart failure [57].
Their prescription needs to be weighed up with the strength of indication. For example all group 2 patients should be on beta-blockers as optimisation of their left sided heart failure [58].
One problematic situation is propranolol used as secondary prophylaxis for oesophageal varices due to liver cirrhosis. Intuitively risk-benefit analysis would point towards treatment (ie. prevention of life-threatening haemorrhage) but evidence from a 10 patient prospective, uncontrolled study in PH associated with portal hypertension (group 1.4.3) showed that withdrawal of beta-blockers improved both functional status and cardiac output without adverse incident [59]. There were no variceal bleeds off beta-blockers over 1½ years, although 60 % with large varices at time of endoscopy were prophylactically banded before discontinuation.
Anti-arrhythmics
Restoration of sinus rhythm (whenever possible) is the best therapeutic strategy for atrial arrhythmias. This is due to the optimisation of haemaodynamics and is especially true for congenital heart disease patients given how poorly tolerated atrial arrhythmias can be. Hence amiodarone, electrical cardioversion and/or electrophysiology studies with ablation are frequently used.
Atrial fibrillation appears to be the most malignant and treatment resistant. One retrospective study of 231 patients with pulmonary arterial hypertension and medically treated CTEPH over 6 years identified 31 episodes of atrial tachyarrhythmia. Sinus rhythm was restored in all atrial flutter patients (n of 12, 3 of which were ablated) but in only 2 out of 13 atrial fibrillation.
Cumulative mortality was 82 % in rate controlled atrial fibrillation compared with 6.3 % when sinus rhythm was restored [60]. Contrary to left heart disease, ventricular arrhythmias appear to be less problematic.
The ESC/ERS guidelines do state digoxin “may be considered” as a rate-controlling strategy for atrial tachyarrhytmias [47].
Diuretics
Over half of all pulmonary hypertension patients are prescribed diuretics and there is undoubtedly symptomatic improvement in the context of right ventricular failure and fluid overload. There is no evidence behind their usage and RCTs will not be forthcoming due the ethics of a placebo arm.
Consequently current best practice, as suggested by the ESC guidelines, is an individualised, clinical decision in the hands of the physician regarding choice, dose and timing of any diuretic therapy, taking into account renal function and serum potassium level [47].
Oxygen
The best evidence for supplemental oxygen therapy comes from the hypoxaemic, group 3 patient population, predominantly COPD. Long term oxygen therapy (LTOT) was proven to confer a survival advantage in hypoxaemic COPD (especially with signs of PH) patients over 30 years ago [61, 62].
In COPD associated PH, LTOT has been shown to improve mean PA pressure at repeat right heart catheterisation by 4.1 mmHg over 19 months [63]. It does appear that approximately only 60 % respond to oxygen therapy and this group has a marked mortality benefit (88 % 2 year survival compared with 22 %) [64].
Hence the ESC guidelines for all PH suggest following the COPD guidelines (most recently reaffirmed by NICE in 2010 [65, 66], namely long term oxygen therapy (used for at least 15 h a day) when resting paO2 is less than 8.0 kPa [47].
40 % of PAH patients in the REVEAL registry were on LTOT [55]. Nocturnal desaturation is likely to be common even in the IPAH group (77 % in this 2001 study of 13 patients, predicted by resting paO2, exertional desaturation and impaired spirometry/gas transfer) [67]. Therefore even in the absence of sleep disordered breathing, consideration should be given to overnight oximetry. Nocturnal oxygen therapy in PH has not been studied (except for Eisenmenger’s syndrome) but keeping oxygen saturations above 90 % seems sensible.
Exercise
There is a growing body of evidence that supervised rehabilitation programmes are beneficial in PH. The putative mechanism is not dissimilar to skeletal muscle weakness as in COPD. First examined by prospective cross-over trial in 2006 showed a 22 % improvement in 6 min walk test after 15 weeks. There was also statistically significant improvement in functional class, quality of life questionnaires and VO2 max. Due to safety concerns the exercise regimen was commenced whilst participants were hospital inpatients but there were no adverse events witnessed. There was not any difference in echocardiographic measurements [68].
Specific Aetiologies
Anorexigen Induced (Nice Group 1.3)
Several of the targeted vasodilator studies included PAH secondary to anorexigen usage and it seems to behave in a not dissimilar fashion to IPAH [72] and hence its treatment follows suit. One difference is a slightly higher response rate to vasoreactivity testing (and hence calcium channel blocker use), 13.4 % in one study [73].
Connective Tissue Disease (Nice Group 1.4.1)
As above, connective tissue disease (CTD) associated PH was often included in the targeted vasodilator studies and hence its treatment algorithm mirrors that of IPAH [47]. It is however generally believed that the CTD cases respond less well to vasodilators than IPAH and often subgroup analyses of the above landmark trials failed to reach significance.
The strongest body of evidence comes from endothelin receptor antagonists and this uncontrolled trial of bosentan in all CTD PH revealed an improvement in functional class in 27 % [74].
There is one RCT with system sclerosis which showed an improved 6MWT on intravenous epoprostenol [75].
Systemic lupus erythematous and mixed connective tissue disorder patients with milder PH (FC I, II or III with a cardiac index of more than 3.1 l/mi/m2) appear to benefit from immunospressive therapy alone (cyclophosphamide and corticosteroids) [76].
Although there is a degree of vasoreactivity at right heart catheterisation, CTD related PH patients do not respond longer term to calcium channel blockers and consequently their use should be avoided. One postulated mechanism is the increased incidence of veno-occlusion, giving a PVOD like picture [73, 77]. Care needs to be given to anticoagulation due to the higher incidence of bleeding diatheses.
Human Immunodeficiency Virus (Nice Group 1.4.2)
Patients with Human Immunodeficiency Virus (HIV) have an increased risk of PAH development which increases their morbidity and mortality. The pathogenesis may involve a direct insult from the virus itself causing endothelial dysfunction in the lung vasculature, with the HIV Nef protein implicated in several studies [78]. PAH can develop in patients with well controlled HIV infection, on or off antiretroviral therapy.
Patients with confirmed HIV associated PAH are treated with antiretroviral therapy and stabilisation or improvement in pulmonary haemodynamics has been reported [79–81]. In the majority of cases advanced pulmonary vasodilator therapy is also commenced following diagnosis. This decision is made according to the patient’s symptoms and other prognostic factors including their PVR, with careful prospective monitoring to ensure treatments are initiated and escalated when appropriate. Patients in WHO functional class III and IV are routinely offered both therapies at diagnosis.
Drug interactions between antiretroviral therapy and advanced pulmonary vasodilator therapies are important in patient management. HIV protease inhibitors mediate Cytochrome P450 isoenzyme inhibition. This markedly increases the levels of phosphodiesterase inhibitors as they are metabolised by Cytochrome P450 [82]. Sildenafil is mainly metabolised by Cytochrome P450 and a safe and effective dose for its concurrent use with protease inhibitors has not been established and its use is therefore contraindicated. If used, therapeutic drug monitoring is necessary [83]. The US Food and Drug Administration advise that when patients are taking protease inhibitors to instead start tadalafil when clinically necessary at a reduced dose of 20 mg once daily, increased to 40 mg once daily based upon individual tolerability.
Endothelin receptor antagonists (ERA) are also metabolised in the liver by Cytochrome P450 and therefore when prescribed with protease inhibitors, elevated ERA plasma concentrations can result [84, 85]. When prescribing bosentan in combination with protease inhibitors dose reductions are therefore recommended at 62.5 mg once daily or every other day [86]. There is less potential for increased ambrisentan levels when taking protease inhibitors and therefore only careful clinical monitoring is deemed appropriate with no dose change necessary. Co-administration of bosentan and the protease inhibitor atazanavir (Reyataz) without ritonavir is not recommended as the level of atazanavir is significantly reduced by the Cytochrome P450 isoenzyme inductive effects of bosentan (unlike ambrisentan) [87]. This has not been demonstrated with other protease inhibitors.
Bosentan has been successfully used as advanced pulmonary vasodilator therapy in HIV associated PAH [81, 88, 89] including 5 % of patients in the EARLY study bosentan treatment arm [11]. Other pulmonary vasodilator therapies that have been successfully used in the treatment of HIV associated PAH include intravenous epoprostenol [90–92], subcutaneous trepostinil [93] and inhaled iloprost [94]. Significant drug interactions with antiretroviral agents are not anticipated with these therapies. The use of combination therapies in patients with HIV associated PAH have only been published as single cases [81] and in positive RCTs inclusive of but not powered to investigate HIV subpopulations [38, 41, 42].
Data relating to the treatment of HIV associated PAH is limited and the management of patients is not well established. Treatment choices are influenced by the potential interactions between advanced pulmonary vasodilator treatments and co-administered anti-retroviral agents. Especially at the beginning of combined treatment, patients are carefully monitored for evidence of drug toxicity, with regular clinical review. In our current practice we routinely commence tadalafil as first line targeted treatment in HIV associated PAH, at the lower dose advised if on a concurrent protease inhibitor. If dual advanced pulmonary vasodilator treatment is appropriate we would then commence an ERA at the advised dose.
Portal Hypertension (Nice Group 1.4.3)
Pulmonary hypertension secondary to liver disease occurs only in the context of portal hypertension. The overall prevalence of PAH in portal hypertension is 2 % [95] and up to 8.5 % in the most severely affected population awaiting liver transplantation [96]. It is important to note that porto-pulmonary hypertension is a very distinct clinical entity from hepatopulmonary syndrome which is caused by microscopic arteriovenous malformations and lowers PVR.
The exact pathophysiology of porto-pulmonary hypertension has not been elucidated but is believed to be due one or more vasoactive cytokines (for example endothelin-1) bypassing their usual metabolism in the liver via porto-systemic shunting and directly impacting on the pulmonary vascular bed [97]. Histological findings are identical to IPAH. In addition to vasoconstriction, contributing factors likely include smooth muscle proliferation, in situ thrombosis, thromboembolism and a hyperdynamic circulation.
The same IPAH treatment algorithm for advanced therapy is followed but the evidence basis is less strong [47]. There are observational studies with each class of advanced vasodilator showing efficacy [98–100]. Consideration needs to be given to the vasodilatory impact on the portal circulation with prostacyclin analogues reported to cause increased flow and progressive splenomegaly [101]. Despite the adverse effect of hepatic dysfunction with endothelin receptor anatgonists, they do appear safe to use [102].
One difference is that vasoreactivity testing need not be performed as calcium channel blockers would not be used due to their effect on systemic vascular resistance. Anticoagulation is not advised due to the coagulopathy and the (especially variceal) bleeding risk. Beta blockers are discussed above.
Historically porto-pulmonary hypertension was considered an absolute contra-indication to liver transplantation. This is no longer the case, especially with mild disease, and there are case reports of PH resolving post-transplantation [103]. However outcomes are worse with more advanced PH [96]. Advanced vasodilators (usually intravenous prostanoids) are frequently employed in the perioperative period.
In terms of patients with co-existent liver dysfunction and pulmonary hypertension of another cause, care is needed with the choice of targeted vasodilator. Ambrisentan is preferred over bosentan but a PDE5 inhibitor would be first-line. Close monitoring of liver function tests is necessary.
Congenital Heart Disease (Nice Group 1.4.4)
Most severely affected (and well-studied) in congenital heart disease associated PH are those in whom Eisenmenger’s syndrome has developed (elevated pressures causing reversal of flow via intra-cardiac shunt or patent ductus arteriosis). Historically considered untreatable, vasodilators have shown to improve clinical and haemodynamic outcomes [104].
Bosentan carries the most weight of evidence. Some non-Eisenmenger congenital heart disease patients were included in the EARLY trial [11]. The BREATHE-5 trial showed good efficacy of bosentan in terms of function and haemodynamics with FC III over 16 and 40 weeks (placebo RCT and open label respectively) [105]. In addition, by having similar impact on pulmonary and systemic vascular resistance, there was no worsening of shunt and hence oxygenation was maintained.
There is unblinded prospective evidence for sildenafil that it can also improve oxygenation levels as well as functioning [106, 107].
Advanced vasodilators also carry a reduction in all-cause mortality as seen with the Royal Brompton Hospital’s cohort of 229 Eisenmenger’s patients. The majority were talking bosentan and were followed-up for several years [108].
Anticoagulation is an individualised risk-benefit decision given both thrombotic tendency (reduced pulmonary flow, secondary erythrocytosis) and haemorrhagic complications (haemoptysis, cerebrovascular) seen in this population. Calcium channel blockers should not be prescribed as they can worsen the right to left shunt with disastrous consequences. Despite being contrary to common sense, nocturnal oxygen does not appear to have any effect [109] and long term oxygen therapy is only recommended if there is improvement if terms of symptoms and oxygenation. Aggressive correction of arrhythmias is particularly important.
Schistosomiasis (Nice Group 1.4.5)
Schistosomiasis may be the commonest cause of PH worldwide but, other than antiparasitic agents, its best treatment is completely unknown. It is caused by infection with the trematode (blood fluke) parasite Schistosoma and affects over 200 million people worldwide. 7.7 % of those with chronic hepatosplenic schistosomiasis have pulmonary hypertension at RHC [110]. Transmission is through contaminated water with freshwater snails the intermediate hosts and it is endemic in wide areas of Africa, Asia, the Middle East and South America.
Of the three commonest species of the parasite, two especially (Schistosoma mansoni and japonicum) cause pulmonary hypertension. The larval form enters through the skin and travel through the circulation to the liver and intestine. After 2 months’ maturation, eggs are produced and deposit in the pulmonary vascular bed, both causing mechanical obstruction and a granulomatous endarteritis through inflammatory cytokine secretion. Portal hypertension also contributes to the pathophysiology.
Given the high prevalence of schistosomiasis and the likelihood of re-infection post treatment, public health strategies have focused more on prevention, especially the provision of clean water and snail control. Praziquantel is the first-line antihelminthic and effective as a single dose, however does not appear to improve the haemodynamics [110]. The advanced vasodilators have not been well studied and given the global distribution of the disease and the large costs entailed it does unfortunately seem unlikely that they will be in the near future. There is some very limited evidence (essentially case reports) behind sildenafil’s use [111, 112]. One uncontrolled, observational study on 12 patients using either phosphodiesterase-5 inhibitor or endothelin receptor antagonist showed a FC and 6MWT improvement [113].
Pulmonary Veno-occlusive Disease (Nice Group 1’)
Vasodilators carry a higher risk in pulmonary veno-occlusive disease (PVOD) as being selective for the arterial side there is the chance of pulmonary oedema developing secondary to hydrostatic capillary pressure. There have been fatalities reported with IV epoprostenol [114]. In specialised centres, prostanoids may be commenced safely when done very cautiously and they appear to be efficacious [115, 116].
There is no data with endothelin receptor antagonists or phosphodiesterase-5 inhibitors. Warfarin, oxygen and early referral for lung transplantation are usually considered. There are case reports of immunosuppressive medication (in this case steroids and azathioprine) being used in the context of autoimmune features [117].
Left Heart Disease (Nice Group 2)
The primary therapy of group 2 PH is optimisation of the heart failure [58]. This is the same for heart failure with reduced and preserved left ventricular systolic function and secondary to valvular disease. Reducing left ventricular end-diastolic pressure reduces the passive transmitted PH. ACE inhibitors and beta blockers are the mainstay and neither are contraindicated in PH.
There have been a number of trials of advanced vasodilators in heart failure without much benefit seen and significant adverse events [118, 119]. Currently they are not recommended but trials are ongoing. Sildenafil carries some supporting data predominantly in terms of improved haemodynamics [120, 121].
Chronic Obstructive Pulmonary Disease (Nice Group 3.1)
The mainstay of treatment for all Dana point group 3 patients (lung diseases and/or hypoxia) is, as the name implies, oxygenation. The evidence for oxygen in COPD associated PH is discussed in the previous section. In summary there is improvement of PA pressures but not reversal to normality.
Early observational evidence suggested a role for targeted vasodilators in PH secondary to lung disease, sildenafil especially [122]. However they are of limited use due to the deleterious side-effect of worsening oxygenation. Despite acutely improving haemodynamics, they have the significant downside of worsening of ventilation/perfusion mismatch. This is secondary to deregulation of the hypoxic pulmonary vasoconstriction mechanism. Hence any potential benefit is offset by worsening of the hypoxaemia that is driving the pulmonary hypertension. A cut-off point of FEV1 60 % predicted is sometimes used as a complete contraindication.
In COPD specifically, sildenafil has been shown to improve haemodynamics but this is countered by worsening degree of hypoxaemia [123]. This blinded RCT of sildenafil in COPD did show an improvement in 6MWT [124]. Results for bosentan in COPD have been mixed [125].
Intriguingly, pravastatin has been shown by RCT to improve exercise capacity and pulmonary vascular resistance in COPD associated PH [126]. The proposed mechanism is inhibition of endothelin-1 production.
Severe PH not explained by the severity of lung disease (the recently discarded concept of “out of proportion” disease) is more worthy of advance vasodilator treatment due to the possibility of additional pathophysiological processes. If the mean PA pressure is over 40 mmHg then referral onto specialist centre for consideration of vasodilators may be warranted.
Interstitial Lung Disease (Nice Group 3.2)
The supplemental oxygen guidelines are as those for COPD, although this has not been investigated on a stand-alone basis. Concerns about worsening mismatch with targeted vasodilators are equally prominent.
Sildenafil once again has the most support although the evidence basis is limited. Both sildenafil and epoprostenol acutely lower PVR but gas exchange actually improved with sildenafil maybe by acting more locally and hence enhancing ventilation-perfusion matching [127].
Nebulised iloprost is the preferred prostanoid (with presumed less systemic effects impacting on hypoxic pulmonary vasoconstriction) with demonstrated improvement in function, symptoms and haemodynamics [128]. Intravenous epoprostenol worsens gas exchange and hence increases hypoxaemia.
There is no place for the use of endothelin receptor antagonists. Bosentan is not efficacious in idiopathic pulmonary fibrosis or systemic sclerosis associated ILD as seen by the placebo controlled RCTs [129–131]. Ambrisentan in the ARIES-3 study actually reduced 6MWT by 23 m in ILD associated PH (as it also did to a lesser extent in COPD) [14].
This was supported and made even more concerning by ARTEMIS-IPF which was terminated early after recruiting 492 idiopathic pulmonary fibrosis patients. Of note only 10 % of the study group had PH. The ambrisentan treatment arm showed statistically significant disease progression and more hospitalisation episodes compared with placebo. There was also non-significant higher mortality of 7.9 % compared with 3.7 % (p of 0.10) [9].
Sleep Disordered Breathing (Nice Group 3.4)
Continuous positive airways pressure treating obstructive sleep apnoea has been shown to reduce pulmonary artery pressure in a well-designed cross-over trial using sham CPAP (subtherapeutic positive end-expiratory pressure) as the control arm [132]. Repeat polysomnography on nocturnal CPAP therapy is indicated to ensure the apnoea-hypopnoea index and level of oxygenation is acceptable.
In the context of PH secondary to obesity hypoventilation syndrome then non-invasive ventilation plus/minus oxygen therapy is likely to be needed and beneficial to the PH but has not been directly studied.
Weight loss is highly important but often difficult to achieve. The wide health benefits of bariatric surgery include significant reduction in mean PA pressures [133].
There is no evidence to support the use of targeted vasodilator medication in sleep disordered breathing associated PH.
Chronic Thromboembolic Disease (Nice Group 4)
Pulmonary thromboendarterectomy for chronic thromboembolic pulmonary hypertension (CTEPH) is discussed in the next chapter. There remains a role for advanced vasodilators as a bridge to surgery and in those not amenable to surgical intervention (for example with distal thromboembolic disease or co-morbidities precluding such major surgery) [134].
Medical therapy results in only limited improvement and, unlike surgery, is in no way curative. Bosentan is the best researched and the BENEFiT placebo RCT showed a 22 % reduction in PVR but no benefit in terms of exercise capacity [135]. A 2010 metanalysis of 10 (mainly open label, uncontrolled) bosentan studies showed significant but modest improvements in 6MWT and PA pressure [136]. Ambrisentan increased the mean 6MWT by 17 m and reduced BNP by 22 % in 28 uncontrolled, proximal and distal CTEPH patients [14].
Prostacyclin analogues and phosphodiesterase-5 inhibtors do also have some supporting evidence [137–140]. Riociguat has also been investigated with promising results in this group and more detail is given below.
The use of vasodilators as a bridge to surgery is only done at the severe end of the spectrum with evidence of right ventricular dysfunction. FC III and IV patients with high a PVR (over 15 Wood units) have been tested with intravenous epoprostenol. This revealed haemodynamic improvement and a post-operative mortality of 8.3 % that (although far higher than those with less severe disease) compared favourably with other studies in similarly advanced patients [141]. There is a evidence against the need for routine use pre-operatively [142].
The final patient group in whom medical therapy is considered are those with persisting PH post pulmonary endarterectomy. There is limited evidence but the BENEFiT trial included this group and they comprised 28 % of the cohort [135]. Despite PH persisting at the 3 month mark post-operatively, when advanced vasodilators are commenced, there is no increased mortality for at least 4 years compared with those who responded well [143].
Sickle Cell Disease (Nice Group 5.1)
Previously in group 1, the haemolytic anaemias including sickle cell have been moved to group 5 following the 2013 world symposium in Nice [144]. The results with targeted vasodilators in sickle cell disease related PH have been disappointing. There is pathophysiological rationale for using phosphodiesterase-5 inhibitors, given the lack of NO bioavailability in its pathogenesis, but sildenafil has shown a high rate of complications, specifically vaso-occlusive crises [145].
l-arginine, a precursor of NO, given orally, produces an acute reduction in PA pressures (by a mean of 15 %) [146].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


