and reactive oxygen species.
Ischemic preconditioning (IPC), that is, exposure to short (5-min) episodes of hypoxia and reoxygenation, protects the heart against injury caused by a subsequent prolonged episode of ischemia and reperfusion.
8.1 Coronary Blood Flow
The coronary circulation, named for its crownlike appearance, encompasses arteries, microvessels, and veins that irrigate and drain the myocardium. When a large coronary artery is severely stenosed, the corresponding perfused myocardial region suffers from ischemia (inadequate blood supply)1 and resulting hypoxia 2; a complete luminal obstruction causes oxygen deprivation and acute myocardial infarction.3
The coronary circulation can be subdivided into the epicardial and intramyocardial compartments, both containing arteries and veins and the latter the microvasculature. The left and right coronary arteries and their main epicardial branches run on the surface of the heart. The coronary arterial circuit generates after multiple branchings a dense mural capillary circuit that deliver oxygen and other nutrients to adjoining cardiomyocytes. Deoxygenated blood is collected by the coronary venous drainage that includes the great, middle, and small cardiac veins and anterior cardiac veins.
Heterogeneous flow distribution inside the cardiac wall depends on the architecture of the circuit as well as on perfusion pressure. Literature data demonstrate that metabolic control of flow, vasomotion, microvascular coronary α-adrenoceptor–primed vasoconstriction, oxygen delivery and consumption, glucose uptake, and glycolytic enzyme activity depend on this heterogeneous distribution.
8.1.1 Coronary Arteries
Coronary arteries supply blood to the myocardium (Vol. 6, Chap. 1. Anatomy of the Cardiovascular Apparatus). The major arteries of the coronary circulation are: the left main coronary that divides into left anterior descending and circumflex branches, and the right main coronary artery.
8.1.1.1 Formation of the Coronary Arterial Bed
Formation and maturation of the myocardium begins during embryo- and fetogenesis and continues after birth [595]. The embryonic coronary vessels initially form as a vascular plexus that covers the outer surface of the heart [596].
The primitive embryonic heart is composed of a compact layer of myocardium lined internally by the endocardium. A second trabecular layer of myocardium is then derived from and remains anchored to the outer compact muscular layer, emerging between this compact layer and endocardium [595]. It comprises a set of thin interconnected strands of muscular trabeculae that constitute fingerlike projections into the ventricular chambers. The inner trabecular layer of myocardium undergoes compaction and hence thickens prior to and after birth.
The compact myocardial layer of the embryonic heart is irrigated by a fetal coronary circulation with an intramyocardial bed. On the other hand, the trabecular layer receives oxygen by diffusion from cardiac chamber blood [595, 596]. During the perinatal period (compaction), the trabecular myocardial layer condenses and coalescence of the trabecular with compact myocardium traps a subset of endocardiocytes that build a vascular plexus and then new coronary vessels. This new layer of compact myocardium thus becomes richly supplied with coronary vessels. The endocardial-to-coronary endothelial lineage conversion occurs within a brief period after birth at least in mice [596]. The vasculature of the inner myocardium is thus not achieved by angiogenesis from the preexisting embryonic and fetal coronary network [596]. At least in adult mouse hearts, 60 % of the myocardium is irrigated by vessels formed from endocardiocytes during trabecular muscle compaction.
The majority of endotheliocytes of inner wall vessels originate from endocardiocytes [596]. The postnatal construction of the inner myocardial vasculature relies on hypoxia and vascular endothelial growth factor, which control the development of the fetal coronary circuit. This generation of coronary vessels in the inner wall also establishes the circulation of the muscular interventricular septum that forms prior to birth also by compaction.
The transition from fetal to postnatal circulation acutely increases the work of the left ventricle. The creation of the coronary vasculature from endocardium enables a rapid vascular and myocardial growth and hence it can matchthe rapidly increasing metabolic demands of the thickening myocardial wall more efficiently than the slower angiogenic expansion of the fetal coronary circulation [596].
8.1.1.2 Architecture of the Coronary Arterial Bed
In general, two coronary left and right arteries originate from coronary ostia at the root of the aorta. More precisely, the coronary ostia localize to the left and right Valsalva sinuses, respectively, just above the closed aortic valve, that is, at the level of ends of open aortic valve cusps. They then emerge between the pulmonary trunk and left and right auricle, respectively.
The left and right coronary arteries divide into primary relatively large epicardial branches. In turn, these branches give rise to parietal arteries that penetrate perpendicularly the epicardium and arterioles that progress inward to cross the myocardium and form subendocardial arterial plexus.
Small penetrating arteries (diameter 100–500 μm) traverse the cardiac wall to deliver blood from the epicardial arteries to the subendocardial microvasculature. A substantial pressure drop occurs in penetrating arteries.
Arterioles (diameter 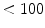 μm) are the primary sites of vascular resistance. Coronary arterioles branch into numerous capillaries that run adjacent to cardiomyocytes. A high capillary-to-cardiomyocyte ratio, and hence a short diffusion distance, ensures adequate oxygen delivery to cardiomyocytes and removal of wastes from them.
μm) are the primary sites of vascular resistance. Coronary arterioles branch into numerous capillaries that run adjacent to cardiomyocytes. A high capillary-to-cardiomyocyte ratio, and hence a short diffusion distance, ensures adequate oxygen delivery to cardiomyocytes and removal of wastes from them.
μm) are the primary sites of vascular resistance. Coronary arterioles branch into numerous capillaries that run adjacent to cardiomyocytes. A high capillary-to-cardiomyocyte ratio, and hence a short diffusion distance, ensures adequate oxygen delivery to cardiomyocytes and removal of wastes from them.The right coronary artery irrigates the right atrium and ventricle as well as the posterior wall of the left ventricle, thereby supplying the sinoatrial node and conus arteriosus. It can then anastomose with the left coronary artery.
The marginal branch descends to the apex along the right margin and supplies the main part of the anterior wall of the right ventricle. The posterior interventricular branch runs in the posterior interventricular sulcus toward the apex and supplies the diaphragmatic regions of both ventricles and posterior one-third of the interventricular septum as well as the atrioventricular node.
The left coronary artery supplies the left atrium and the lateral and anterior walls of the left ventricle. It divides into the anterior interventricular branch and the circumflex artery. The former descends to the cardiac apex in the anterior interventricular sulcus and yields the main supply to the anterior regions of both ventricles and anterior two-thirds of the interventricular septum. The latter anastomoses with the right coronary artery.
The sinoatrial node is often perfused by the left coronary artery rather than the right one. The atrioventricular node is occasionally irrigated by the left coronary artery.
8.1.1.3 Between-Cardiac Cavity Distribution of the Coronary Blood Flow
At rest, the right ventricular blood flow per unit mass (expressed in gram) of myocardium is typically 50–60 % of the left ventricular blood flow, at least in dogs and horses [597]. In resting swine, the right ventricular blood flow per unit mass of myocardium is about 70–90 % of the left ventricular blood flow. The lower resting flow in the right ventricle can be explained by a smaller wall thickness and oxygen consumption than those of the left ventricle.
During graded treadmill exercise in dogs, horses, and pigs, the right ventricular blood flow increases proportionally to the cardiac frequency. The right ventricular blood flow per unit mass of myocardium most often equals 75–90 % of the left ventricular blood flow during maximal exercise [597].
At rest, the right and left atrial blood flows per unit mass of myocardium are generally 20–40 % of the left ventricular blood flow in dogs, horses, and pigs [597].
During treadmill exercise, atrial flows can increase up to 15-fold reaching 50–60 % of the left ventricular flow during heavy exercise in dogs and swine, and up to 70 % in ponies [597]. Within the atria, blood flow is the lowest in the appendages, but the exercise-induced increase in blood perfusion is greater in the appendages.
8.1.1.4 Anatomical Coronary Arterial Variations
Occasionally, the left and right coronary arteries arise from a common trunk. On the other hand, three coronary arteries can originate from the aortic root. In addition, the posterior interventricular branch usually arises from the right coronary artery, but sometimes (∼ 10 %) from the left coronary.
Alternative architectures of the coronary arterial circuit determine dominance of the right or left coronary artery. The artery that supplies the posterior descending artery determines the coronary dominance. The right and left dominance correspond to a main perfusion by the right coronary and circumflex artery, respectively. Codominance refers to as similar supply by both coronary arteries.
8.1.1.5 Coronary Arterial Anomalies
Some congenital coronary anomalies have no clinical repercussion. They are diagnosed fortuitously. On the other hand, major coronary anomalies have clinical impact due to resulting ischemia or volume overload (i.e., angina, dyspnea, arrhythmia, and sudden death).
Major coronary anomalies detected in children and adults by transthoracic echocardiography and coronaroangiography include [598]: (1) abnormal origins of coronary artery from the opposite sinus with interarterial course; (2) anomalous left coronary artery from the pulmonary artery; (3) single coronary ostia; and (4) coronary fistula.
8.1.1.6 Coronary Arterial Anastomoses
Intracardiac anastomoses between branches of the two coronary arteries can exist. Moreover, extracardiac anastomoses between coronary arteries and arteries of neighboring organs can be detected. In particular, they give rise to periadventitial arteries of the ascending aorta and other thoracic vessels, especially vasa vasorum of the ascending aorta and pulmonary artery. These extracardiac branches of the coronary arteries emerge commonly from regions near the aortic root, the base of the pulmonary arterial trunk, pulmonary veins, and ostia of the superior and inferior venae cavae, that is, at reflections sites of the pericardium that occur during development at arterial and venous ends of the heart [599]. These extracardiac anastomoses connect to pericardial, mediastinal, diaphragmatic, intercostal, and bronchial arteries, thereby irrigating up to the pulmonary hila, trachea, and esophagus. The largest communication is that with the pericardiacophrenic branches of the internal mammary arteries traveling with the phrenic nerves.
Recently, the most common anastomoses were found from bronchial and internal thoracic arteries to coronary arteries rather than from pericardiacophrenic branches of the internal thoracic arteries [600]. To a lesser extent, anastomoses exist from anterior mediastinal, intercostal, and esophageal arteries. These observations can result from interindividual anatomical variability.
8.1.1.7 Collateral Coronary Arterial Circulation
The collateral arterial circulation of the heart constitutes natural bypasses, thereby yielding an alternative source of blood supply to a ischemic myocardial region. Coronary collaterals arise from vasculo-, angio-, and arteriogenesis (Vol. 5, Chap. 10. Vasculature Growth). They can prevent signs of myocardial ischemia during brief vascular occlusions. However, ischemic preconditioning, rather than recruitment of collateral vessels, most often heightens tolerance against myocardial ischemia [601].
The measurement of aortic and intracoronary pressure or velocity using pressure or Doppler sensor-tipped angioplasty guidewires enables the assessment of the pressure- or velocity-derived collateral flow index (CFI) that expresses the amount of flow through collaterals to the perfused region of interest as a fraction of the flow via the normally patent vessel. Collateral flow sufficient to prevent myocardial ischemia during coronary occlusion amounts to at least 25 % of the normal flow through the open vessel [601].
The pressure-derived CFI is determined by simultaneous measurement of mean aortic (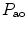 ), mean distal coronary occlusive (
), mean distal coronary occlusive (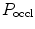 ), and central venous (CVP) pressures [601]:
), and central venous (CVP) pressures [601]:
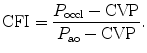
The velocity-derived CFI is measured by distal occlusive coronary flow velocity (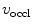 ) and coronary flow velocity in nonoccluded artery (v) taken at the same location and following occlusion-induced reactive hyperemia:
) and coronary flow velocity in nonoccluded artery (v) taken at the same location and following occlusion-induced reactive hyperemia:
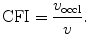
In addition to arterioarterial anastomoses, generation of new arteries (angiogenesis) and enlargement of small arteries (arteriogenesis or collateral growth) contribute to the formation of collateral arterial circuits primed by ischemia. Angiogenesis is related to capillary generation and growth launched by ischemia, but as ischemia wanes, capillary growth can stop and even rarefaction can occur [603]. On the other hand, arteriogenesis continues as ischemia is resolved. Hence, a continuous arterial circuit remodeling can be maintained by mechanical forces.
), mean distal coronary occlusive (), and central venous (CVP) pressures [601]:(8.1)
) and coronary flow velocity in nonoccluded artery (v) taken at the same location and following occlusion-induced reactive hyperemia:(8.2)
The construction of coronary collaterals relies on proliferation of vascular smooth muscle and endothelial cells initiated by growth factors liberated by ischemia. Phenotypic switching of vascular smooth myocytes from a contractile to a proliferate phenotype is triggered by activation of proper signaling pathways with available energy.
Mobile and dynamical mitochondria organized along sarcomeres are thus involved in phenotypic switching under constraint resulting from ischemia-associated oxidative stress resulting from excessive production or inadequate neutralization of reactive oxygen species (ROS) [603].
The antioxidant defense eliminates ROS using mitochondrial and cytosolic free radical scavengers and resolves mismatches between outward and inward proton fluxes at the inner mitochondrial membrane by uncoupling proteins. Uncoupling proteins support proton reentry into the mitochondrial matrix, hence reducing ROS production, but also inhibiting ATP synthase.
The Warburg effect refers to as predominant production of energy in cancerous cells from a high rate of glycolysis followed by lactic acid fermentation in the cytosol rather than by a comparatively low rate of glycolysis followed by oxidation of pyruvate in mitochondria in normal cells.
The metabolic switch that favors aerobic glycolysis over mitochondrial oxidative phosphorylation to meet rapid cellular proliferation relies on UCP2 that counteracts P53 [603].
The magnitude of oxidative stress is correlated with the extent of mitochondrial dysfunction and thus with impaired capacity for arteriogenesis. Therefore, improved mitochondrial respiration and antioxidant defense restore at least partly coronary collateral growth arteriogenesis via AMPK signaling, as AMP/ATP ratio increases under cellular stress [603]. According to the energy status, AMPK enables or prevents phenotypic switching and hence growth and proliferation.
The AMPK kinase modulates cellular energy flux in response to an increased AMP/ATP ratio under stress. It is activated by AMP binding and phosphorylation by LKb1 and CamK kinases, in addition to AMP binding. According to the energy status, AMPK enables or prevents phenotypic switching and hence growth and proliferation.
Coronary collaterals can undergo both short- (i.e., vasomotor tone adaptation) and long-term (i.e., recruitment and growth of collateral vessels) adjustments in response to arterial occlusion.
Mature coronary collaterals are responsive to vasodilators (e.g., nitric oxide (NO) and atrial natriuretic peptide) and vasoconstrictors (e.g., angiotensin-2 and vasopressin), in addition to the platelet products serotonin and thromboxane-A . During effort, β-adrenergic activity and endothelium-derived NO and prostanoids exert vasodilatory influences on coronary collateral vessels [597].
. During effort, β-adrenergic activity and endothelium-derived NO and prostanoids exert vasodilatory influences on coronary collateral vessels [597].
. During effort, β-adrenergic activity and endothelium-derived NO and prostanoids exert vasodilatory influences on coronary collateral vessels [597].When coronary collaterals can supply an adequate arterial inflow to meet the perfused myocardium needs, then the magnitude and distribution of blood flow are determined by the autoregulatory response of the microvasculature in the corresponding myocardial region.
When coronary collaterals cannot deliver sufficient arterial inflow, then the perfusion blood flow will be determined by the conductive collateral circuit with a transmural distribution of perfusion similar to that downstream from the arterial stenosis [597].
8.1.2 Coronary Veins
The venous drainage of the heart include:
The coronary sinus also receives the oblique vein of the left atrium, left marginal vein, and the left posterior ventricular vein. It opens into the right atrium.
1.
Small veins that empty directly into the cardiac chambers such as anterior cardiac veins that collect blood from the right ventricle and open into the right atrium, the smallest cardiac veins draining into any of the four cardiac chambers; and
2.
Large veins, that is, the great, middle, and small cardiac veins that empty into the coronary sinus in the coronary groove at the posterior surface of the heart, between the left atrium and ventricle.
Most blood from the left ventricular myocardium drains into the coronary sinus. The anterior cardiac vein receives blood from the right ventricular myocardium. These receiving veins open into the right atrium. Thebesian veins drain a small proportion of coronary blood directly into the cardiac chambers, hence creating a shunt.
8.1.3 Coronary and Cardiac Innervation
The heart is innervated by autonomic and sensory nerves of the sympathetic trunks and vagus nerves. Postganglionic parasympathetic and sympathetic fibers innervate the nodes and routes of action potential triggering and conducting (nodal) tissue and coronary vessels.
Preganglionic sympathetic nerves from the spinal cord (T1–T6) synapse in cervical and thoracic sympathetic chain ganglia. Postganglionic fibers travel in cervical and thoracic cardiac branches of the sympathetic trunk.
Preganglionic parasympathetic fibers run in cervical and thoracic cardiac branches of the vagus nerves to synapse with ganglion cells of the cardiac plexi near the heart.
Sensory fibers carrying information on blood pressure and flow and cardiac frequency travel in the vagus nerves. Pain fibers are associated with the sympathetic trunks and enter the spinal cord by dorsal roots (T1–T5).
8.1.4 Coronary Artery Physiology
The resting coronary blood flow equals about 250 ml/min (0.8 ml/min/g of myocardium [0.5–1.5 ml/min/g according to the state of alertness [597]]), that is, 5 % of cardiac output. Coronary blood flow occurs mainly during diastole. It is mainly determined by the local oxygen demand.
Exercise and pregnancy require hemodynamic adjustments, effort being the stronger physiological stimulus of increased cardiac output and arterial pressure.
Myocardial oxygen extraction is very high in basal conditions. Any additional metabolic demand thus requires an elevated myocardial blood supply. At rest, the myocardium extracts about 75 % of the oxygen delivered by coronary blood flow [604]. Only a small extraction reserve is available when myocardial oxygen consumption is augmented during exercise. Control mechanisms based on local metabolic feedback and sympathetic β-adrenoceptor–mediated feedforward arteriolar vasodilation fit coronary blood flow to myocardial oxygen consumption.
A further increase in coronary blood flow can be elicited with a pharmacological or ischemic vasodilatory stimulus. A reactive hyperemia is observed after a brief total coronary occlusion during maximal exercise in dogs and pigs. Intravenous administration of adenosine to swine provokes a 15–26 % increase in myocardial blood flow during maximum exercise and about 20 % further decrease in coronary arterial resistance [597]. The coronary vascular resistance is commonly given by the mean aortic pressure/mean coronary blood flow ratio.
The pharmacologically induced increase of coronary blood flow during exercise can enhance cardiac contractility ( Gregg effect). Intracoronary administration of a selective α1-adrenergic blocker during exercise in dogs engenders a 21 % augmentation of coronary artery blood flow and maximal rate of regional myocardial segment shortening (unchanged total systolic shortening) and 26 % elevation of myocardial oxygen consumption [597]. The heightened velocity of shortening does not depend on β-adrenergic activation, as a similar response occurs after blockade of the action of adrenaline and noradrenaline on both β1- and β2-adrenergic receptors. Similarly, intracoronary administration of adenosine during exercise causes a 25–30 % increase in coronary blood flow and 27 % augmentation of the rate of systolic segment shortening and 16 % elevation of myocardial oxygen consumption without change in cardiac frequency, left ventricular systolic pressure, and myocardial end-diastolic segment length [597].
The coronary flow reserve (CFR; the ratio of maximum to basal coronary flow) refers to the capacity of the coronary circulation to increase blood flow using vasodilators, such as nitric oxide and prostacyclin. The vasodilatory reserve during exercise in atrial cavities is comparable to that in the ventricular chambers.
Hemodynamic stress and chemical messengers, such as acetylcholine, bradykinin, and histamine, are the main triggers of nitric oxide release by the endothelium. Flow-induced lectin–oligosaccharide complex formation participates in sensing shear stress applied on the wetted (luminal) endothelial surface. Flow-sensitive and lectinic substances at this surface include, at least in guinea pig hearts [605]:
The amplitude of receptor-induced vascular responses upon receptor activation depends on flow and glycosylation (hyaluronate glycosaminoglycan).
1.
Certain cell adhesion molecules, such as integrins and selectins;
2.
Some G-protein-coupled receptors of adenosine (A –A
–A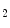 ), angiotensin-2, bradykinin (B
), angiotensin-2, bradykinin (B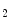 ), endothelin-1, noradrenaline (α1-, β1-, and β3-adrenoceptors), prolactin, thromboxane-A
), endothelin-1, noradrenaline (α1-, β1-, and β3-adrenoceptors), prolactin, thromboxane-A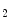 ; and
; and
–A), angiotensin-2, bradykinin (B), endothelin-1, noradrenaline (α1-, β1-, and β3-adrenoceptors), prolactin, thromboxane-A; and3.
Certain receptor protein Tyr kinases such as insulin receptor.
Carbon dioxide, the most potent respiratory stimulant, is a vasodilator for cerebral and myocardial blood vessels. Elevated systemic 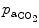 augments blood flow independently from local metabolic demands, engendering an excessive perfusion similarly to that induced by pharmacological vasodilators. In healthy individuals, voluntary breathing maneuvers modify myocardial oxygenation via CO
augments blood flow independently from local metabolic demands, engendering an excessive perfusion similarly to that induced by pharmacological vasodilators. In healthy individuals, voluntary breathing maneuvers modify myocardial oxygenation via CO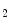 . Apnea and hyperventilation change the blood gas content; capillary
. Apnea and hyperventilation change the blood gas content; capillary 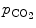 diminishes significantly during hyperventilation and capillary
diminishes significantly during hyperventilation and capillary 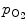 declines markedly during 2-min breathhold [606]. Long breathholds prime CO
declines markedly during 2-min breathhold [606]. Long breathholds prime CO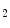 -mediated increase in blood flow, whichcan compensate for O
-mediated increase in blood flow, whichcan compensate for O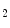 desaturation. Mild hypercapnic hypoxia can further raises the myocardial blood flow, as hypercapnia and hypoxia act synergistically.
desaturation. Mild hypercapnic hypoxia can further raises the myocardial blood flow, as hypercapnia and hypoxia act synergistically. 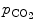 -dependent vasodilation or vasoconstriction determines myocardial oxygenation.
-dependent vasodilation or vasoconstriction determines myocardial oxygenation.
augments blood flow independently from local metabolic demands, engendering an excessive perfusion similarly to that induced by pharmacological vasodilators. In healthy individuals, voluntary breathing maneuvers modify myocardial oxygenation via CO. Apnea and hyperventilation change the blood gas content; capillary diminishes significantly during hyperventilation and capillary declines markedly during 2-min breathhold [606]. Long breathholds prime CO-mediated increase in blood flow, whichcan compensate for O desaturation. Mild hypercapnic hypoxia can further raises the myocardial blood flow, as hypercapnia and hypoxia act synergistically. -dependent vasodilation or vasoconstriction determines myocardial oxygenation.Oxygenation-sensitive cardiovascular magnetic resonance imaging using blood oxygen level-dependent (BOLD) effect in T2 -weighted imaging sequences (
-weighted imaging sequences (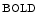 CMRI)4 shows that a 2-min breathhold causes a deoxygenation in the left ventricular blood associated with an increased (8.2 %) myocardial oxygenation [606]. On the other hand, a 2-min hyperventilation that generates coronary vasoconstriction provokes a significant (7.5 %) drop in
CMRI)4 shows that a 2-min breathhold causes a deoxygenation in the left ventricular blood associated with an increased (8.2 %) myocardial oxygenation [606]. On the other hand, a 2-min hyperventilation that generates coronary vasoconstriction provokes a significant (7.5 %) drop in 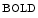 CMRI signal intensity and hence in myocardial oxygenation, deoxygenation of the myocardium resulting from an O
CMRI signal intensity and hence in myocardial oxygenation, deoxygenation of the myocardium resulting from an O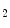 extraction outweighing coronary O
extraction outweighing coronary O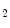 supply even at constant myocardial workload.
supply even at constant myocardial workload.
-weighted imaging sequences (CMRI)4 shows that a 2-min breathhold causes a deoxygenation in the left ventricular blood associated with an increased (8.2 %) myocardial oxygenation [606]. On the other hand, a 2-min hyperventilation that generates coronary vasoconstriction provokes a significant (7.5 %) drop in CMRI signal intensity and hence in myocardial oxygenation, deoxygenation of the myocardium resulting from an O extraction outweighing coronary O supply even at constant myocardial workload.8.1.4.1 Interference Between Myocardium and Coronary Vasculature
Cardiomyocytes represent approximately 75 % of total cardiac volume, but their number accounts for less than 40 % of all cardiac cells (fibroblasts, endotheliocytes, vascular smooth myocytes, macrophages, and circulating blood cells).
The myocardium is irrigated by the coronary vasculature. Coronary perfusion ensures cardiac function. Cardiac function determines myocardial oxygen consumption and consequently coronary perfusion. The latter is regulated by extrinsic factors such as the neurohumoral regulation and intrinsic processes, such as autoregulation.
Autoregulation (Sect. 8.1.4.7) is the intrinsic mechanism by which the coronary arterial circuit maintains a quasiconstant blood flow despite changes in perfusion pressure, excluding influences of extrinsic nerves or bloodborne messengers. It is aimed at matching nutrient input supplied by coronary blood flow to myocardial metabolism.
A mechanical interaction between the contracting myocardium and the coronary vasculature that are in contact explains distribution of the coronary flow in different parietal layers (from subepicardial to subendocardial laminae) and flow variations during the cardiac cycle.
Myocardial contraction–relaxation cycles deform the mural vasculature. During systole with its two phases (isovolumic contraction and ejection), coronary artery inflow is impeded and even reversed; venous outflow is augmented. The magnitude of coronary flow depends on the local transmural pressure. The intramyocardial pressure depends on the local contraction strength as well as the magnitude of isovolumic deformation of the ventricular cavity [607]. During diastole, the collapsing effect of the myocardium on the mural coronary vasculature ceases. The contribution of the myocardium depends on initial myofiber length [607].
Conversely, the coronary vasculature and blood flow affect the myocardium and its contraction. The Gregg effect is related to the observation that an improved coronary perfusion increases myocardial oxygen consumption and contractile function. Positive inotropic factors are released by increased hemodynamic stress. During diastole, an increase in coronary perfusion pressure increases ventricular wall stiffness, but the effect is small [607]. During systole, coronary perfusion affects cardiac contractility by two mechanisms. Increased perfusion pressure increases microvascular volume, thereby opening stretch-activated ion channels that raise cytosolic Ca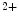 concentration and hence Ca
concentration and hence Ca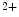 sensitivity, enhancing myocardial contractility (Gregg effect) [607].
sensitivity, enhancing myocardial contractility (Gregg effect) [607].
concentration and hence Ca sensitivity, enhancing myocardial contractility (Gregg effect) [607].Thickening of the shortening myocardium reduces the space allocated to the vasculature. On the other hand, the vascular deformation, and thus vascular emptying that allows myofiber thickening during shortening, enhances myocardial contraction [607]. In other words, when the intramyocardial pump cannot displace intravascular blood, muscle contraction is reduced.
Mechanical crosstalk between the myocardium and coronary vasculature relies on short-term mutually acting mechanical factors. The contracting myocardium that shortens and thickens increases the wall stiffness and ventricular pressure. Elevated vascular filling raises vascular diameters. In addition, the vasomotor tone changes affects the rheology of the vascular wall.
Stunning refers to a transient period of ischemia with hypokinesis and a normal myocardial blood flow rate. On the other hand, sustained limitation of blood flow in myocardial regions perfused by a stenosed coronary artery can lead to a pseudohibernation state in which contractility and metabolism fall to accommodate the persistently reduced perfusion.
8.1.4.2 Myocardial Oxygen Consumption at Rest and During Exercise
The two apposed cardiac pumps continuously eject blood into the systemic and pulmonary circulations. Oxygen consumption per gram of myocardium is 20-fold higher than that of skeletal muscle, which is entirely relaxed with very low metabolic requirements at rest [597]. Cardiac oxygen consumption depends on cardiac chronotropy and inotropy and, hence, on ventricular work.
The heart has the highest oxygen consumption per unit mass among all organs. Cardiac oxygen consumption is mainly related to contraction (10–20 % for basal metabolism [597]. Arterial oxygen extraction ranges from 70 to 80 %, that is, much more than in other organs (25 %).
The high level of oxygen extraction is possible due to a high capillary density of 3000–4000/mm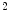 , that is, much higher than that in skeletal muscle (500–2000 capillaries/mm
, that is, much higher than that in skeletal muscle (500–2000 capillaries/mm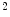 ) [597].
) [597].
, that is, much higher than that in skeletal muscle (500–2000 capillaries/mm) [597].Augmented oxygen demand during exercise, which rises up to sixfold during maximal exercise, is achieved mainly by an increase in coronary blood flow. This adaptation results from acute and chronic regulatory mechanisms that control the coronary blood flow rate. An increase in cardiac frequency decreases the end-diastolic and stroke volume and may account for 50–70 % of the elevation of myocardial oxygen consumption during exercise [597]. The exercise-induced augmentation of contractility results from β-adrenoceptor activation as well as positive inotropic effect of cardiac frequency ( Bowditch–Treppe effect; Vol. 6, Chap. 3. Cardiovascular Physiology). The adrenergically mediated augmentation of contractility is estimated to be 15–25 % of the heightened myocardial oxygen during exercise [597]. Elevated ventricular work is evaluated to account for the remaining 15–25 % of the increase in oxygen consumption [597].
In the right ventricle, oxygen extraction is lower at rest than in the left ventricle and increases substantially during exercise, similarly to skeletal muscles.
In humans, unlike certain mammalian species, oxygen delivery is only slightly facilitated by an elevation of hemoglobin concentrations during exercise. Increased myocardial oxygen demands during exercise are matched chiefly by elevating coronary blood flow due to a combination of coronary vasodilation, decayed coronary vascular resistance (20–30 % from the resting level), and augmented mean arterial pressure (20–40 %) [597]. During effort, coronary blood flow rises in proportion to the cardiac frequency to reach a peak during maximal exercise from three to fivetimes the resting level (7.5–8.5 ml/min/g myocardium for a cardiac frequency 4.3 Hz in swine [597]). However, the increased oxygen extraction indicates that the elevated myocardial blood flow does not fully compensate the increased oxygen demand during exercise.
In young healthy male humans, the coronary venous oxygen content decreases by 8 % and saturation from about 33 % at rest to approximately 24 % during heavy exercise at about 90 % of maximal cardiac frequency, whereas coronary venous oxygen partial pressure was minimally modified [597]). A rightward shift of the hemoglobin oxygen dissociation curve facilitates oxygen delivery to the myocardium during heavy exercise possibly due to a decreased blood pH linked to lactate production by working skeletal muscles.
Energy production in the normally functioning heart depends primarily on oxidative phosphorylation, with less than 5 % of ATP produced from glycolysis. Hence, increase in cardiac activity relies on instantaneous elevation of oxygen availability.
8.1.4.3 Determinants of Coronary Blood Flow
Working skeletal muscles that cyclically compress neighboring veins increase the venous return to the heart, hence stroke volume. Elevated preload and cardiac sympathetic activity increases the cardiac output.
Perfusion Time
An increase in cardiac frequency impinges on diastolic time more than systolic time, hence not only simply reducing the perfusion time but also altering the efficient perfusion period.
Coronary Perfusion Pressure and Resistance
The coronary perfusion pressure is defined as the difference between the time-dependent aortic diastolic pressure and the central venous pressure (CVP; i.e., right atrial pressure).
The coronary arterial bed can be subdivided into two compartments: (1) conductive large and mid-sized arteries (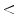 5 % of total coronary resistance) and (2) resistive small arteries (diameter 100–500 μm) and arterioles.
5 % of total coronary resistance) and (2) resistive small arteries (diameter 100–500 μm) and arterioles.
5 % of total coronary resistance) and (2) resistive small arteries (diameter 100–500 μm) and arterioles.In addition, small arteries and arterioles are subjected to interaction between the intravascular distending pressure and the extravascular compressive pressure that tends to collapse parietal vessels during systole.
However, atherosclerosis in large coronary arteries or vasospasm can yield a substantial fraction of total resistance.
Systolic Compression of Parietal Coronary Vessels
However, intraparietal coronary vessels are deformable, especially during systole, during which they are more or less collapsed. Therefore, the coronary perfusion pressure is not only dictated by the pressure drop between the entry and exit of the coronary circuit compartment, but by time- and space-dependent transmural pressure. Coronary blood flow must be characterized by pressure–flow relations over a range of perfusion pressures. The effective pressure drop is the difference between inlet and outlet transmural pressures. However, the external applied pressure which varies both temporally and spatially is unknown.
The coronary pressure–flow relations depend on various factors. Increased cardiac frequency decreases ejection volume and hence coronary systolic input volume, as it reduces the total systolic time. Increased contractility raises systolic compression of intramural coronary vessels. On the other hand, increased myocardial relaxation elevates the diastolic perfusion time. Increased left ventricular diastolic filling pressure attenuates coronary blood flow.
During systole, intramyocardial blood vessels are compressed and twisted by the contracting myocardium, thereby limiting blood flow rate. The greatest throttling effect happens in the subendocardium, generating a transmural distribution of ventricular myocardial blood flow. Furthermore, systolic blood flow in compressed arteries and arterioles in the innermost ventricular wall layer is squeezed retrogradely into subepicardial arteries. Nonetheless, augmented diastolic flow can compensate systolic underperfusion, as the subendocardium has a 10 % higher arteriolar and capillary density [597].
On the other hand, intramyocardial venous blood is propelled forward toward the coronary sinus as well as retrogradely. Hence, upstream and downstream compartments of epicardial vessels are capacitors that store blood.
Anyway, the increase in external compression does not represent a very important factor in the normal coronary circulation, because the coronary vasodilatory reserve capacity persists even during maximal effort, the subepicardial wall layer being much less exposed. Nevertheless, when the coronary blood flow or its oxygen-carrying capacity decays upon hypoxia or anemia, then extravascular forces can significantly limit the coronary blood flow rates during exercise.
Diastolic Compensation
During diastole, when the myocardium relaxes and when the aortic valve leaflets do not impede flow at coronary artery inlets, blood flow rate rises again to reach a peak that has a higher amplitude than the systolic one. Therefore, the bulk coronary irrigation results from a balance between systolic and diastolic events.
Nervous Control of Coronary Arterial Resistance
The control of coronary vascular resistance is carried out by vasodilatory and vasoconstrictory signals transmitted remotely by the autonomic nervous and endocrine systems as well as locally by the endothelium via secreted vasoregulators and the myocardium via metabolic mediators (e.g., vasodilators adenosine, ATP, CO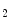 , and H
, and H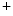 ion).
ion).
, and H ion).α-adrenergic receptors influence coronary blood flow via three mechanisms: (1) prejunctional α2-adrenoceptors and negative feedback of noradrenaline release that prevents cardiac β-adrenoceptor stimulation, both β1- and β2-adrenoceptors stimulating the rate (chronotropy) and force (inotropy) of heart contraction, thereby precluding elevation of heart rate, stroke volume, and systolic pressure; (2) α1- (predominantly) and α2-adrenoceptors on smooth myocytes of small coronary arteries and arterioles that cause vasoconstriction; and (3) α2-adrenoceptors on coronary vascular endothelium that stimulate nitric oxide release.
At rest, the cardiac sympathetic activity is minimal. During effort, α-adrenoceptor–mediated coronary vasoconstriction compete with metabolically induced coronary vasodilation. Postjunctional α1- (principally) and α2-adrenoceptors can augment subendocardial blood flow, hence enhancing the subendocardial/subepicardial blood flow ratio in an healthy coronary bed during exercise [597]. On the other hand, α-adrenoceptors generate a transmurally uniform coronary vasoconstriction, as their blockage increases blood flow uniformly in the entire wall thickness in myocardial regions perfused by a stenotic coronary artery as well as in the pressure-overload–induced hypertrophy of the left ventricular wall of dogs during exercise. Humoral adrenergic activation upon intracoronary noradrenaline injection raises blood flow uniformly in all myocardial layers.
β2-adrenergic receptors reside in coronary arterioles. As for α-adrenoceptor–mediated control, the control exerted by β-adrenergic receptors on the coronary circulation is minimal at rest. In normal human subjects or patients with angiographically normal coronary arteries, β-adrenoceptor blockade by a nonselective β-blocker decreases myocardial blood flow during bicycle exercise to a greater extent than the reduction of myocardial oxygen consumption, β-adrenoceptor activation contributing to coronary vasodilation during exercise via a feedforward mechanism [597].
Intracoronary administration of a nonselective β-adrenergic blocker in exercising dogs causes a slightly greater decrease of coronary blood flow than does a selective β1-adrenergic blocker, as β2-adrenoceptors contribute to adrenergic coronary vasodilation.
The coronary resistive vessels are richly innervated by parasympathetic fibers that engender a coronary vasodilation, as acetylcholine provokes nitric oxide release from the vascular endothelium.
Parasympathetic effects on both the myocardium and coronary arterial bed are negligible during submaximal exercise, as the myocardial vagal tone progressively withdraws during increasing levels of exercise. Vagal tone removal may contribute to β-adrenergic vasodilation at low exercise intensity. However, the global effect of vagal nerve stimulation depends on the mammalian species. For example, the acetylcholine-induced NO-mediated vasodilation, which predominates in dogs, is outweighed in swine by a direct vasoconstrictory effect of acetylcholine on coronary smooth myocytes, resulting in a net vasoconstriction.
Matabolic Control of Coronary Arterial Resistance
Myocardial oxygen and carbon dioxide partial pressures operate synergistically to raise coronary blood flow according to the cardiac frequency. Carbon dioxide dilates coronary arterioles possibly via an acidosis-induced opening of K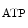 channels. During exercise, coronary CO
channels. During exercise, coronary CO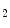 partial pressure and pH remain nearly cosntant.
partial pressure and pH remain nearly cosntant.
channels. During exercise, coronary CO partial pressure and pH remain nearly cosntant.Adenosine that predominantly dilates arterioles is a messenger that adapts coronary resistive vessel caliber to varying myocardial metabolic needs. In normal conditions, adenosine is generated independently of the metabolic state of the cardiomyocyte mainly from extracellular AMP by 5′-ectonucleotidase and from 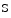 adenosylhomocysteine by
adenosylhomocysteine by 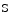 adenosylhomocysteine hydrolase. Cardiomyocytes use newly formed adenosine to form AMP via adenosine kinase. When the cardiac work rises, cytosolic free ADP increases, and adenosine is produced within the cell. Adenylate kinase transforms two molecules of ADP into one molecule of ATP and AMP. The latter is catabolized by AMP 5′-nucleotidase into adenosine. When the cytosolic adenosine concentration increase from the normal level of 0.8–2 μmol, adenosine is degraded by adenosine deaminase or transported out of the cell into the interstitium via nucleoside transporters where it can cause vasodilation possibly via A
adenosylhomocysteine hydrolase. Cardiomyocytes use newly formed adenosine to form AMP via adenosine kinase. When the cardiac work rises, cytosolic free ADP increases, and adenosine is produced within the cell. Adenylate kinase transforms two molecules of ADP into one molecule of ATP and AMP. The latter is catabolized by AMP 5′-nucleotidase into adenosine. When the cytosolic adenosine concentration increase from the normal level of 0.8–2 μmol, adenosine is degraded by adenosine deaminase or transported out of the cell into the interstitium via nucleoside transporters where it can cause vasodilation possibly via A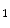 and A
and A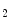 receptors coupled with K
receptors coupled with K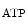 and the cAMP–PKA pathway, in particular A
and the cAMP–PKA pathway, in particular A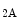 on vascular smooth myocytes [597].
on vascular smooth myocytes [597].
adenosylhomocysteine by adenosylhomocysteine hydrolase. Cardiomyocytes use newly formed adenosine to form AMP via adenosine kinase. When the cardiac work rises, cytosolic free ADP increases, and adenosine is produced within the cell. Adenylate kinase transforms two molecules of ADP into one molecule of ATP and AMP. The latter is catabolized by AMP 5′-nucleotidase into adenosine. When the cytosolic adenosine concentration increase from the normal level of 0.8–2 μmol, adenosine is degraded by adenosine deaminase or transported out of the cell into the interstitium via nucleoside transporters where it can cause vasodilation possibly via A and A receptors coupled with K and the cAMP–PKA pathway, in particular A on vascular smooth myocytes [597].Coronary arteriolar smooth myocytes possess K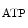 channels responsible for an outward flux of K
channels responsible for an outward flux of K ions that hyperpolarizes the plasma membrane. The subsequent decreased Ca
ions that hyperpolarizes the plasma membrane. The subsequent decreased Ca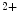 influx causes a vasodilation. Adenosine, prostacyclin, and β2-adrenoceptors increase K
influx causes a vasodilation. Adenosine, prostacyclin, and β2-adrenoceptors increase K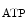 activity via the cAMP–PKA axis; NO activates the K
activity via the cAMP–PKA axis; NO activates the K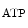 channel via the cGMP second messenger.
channel via the cGMP second messenger.
channels responsible for an outward flux of K ions that hyperpolarizes the plasma membrane. The subsequent decreased Ca influx causes a vasodilation. Adenosine, prostacyclin, and β2-adrenoceptors increase K activity via the cAMP–PKA axis; NO activates the K channel via the cGMP second messenger.Adenosine effect is small and thus does not represent a principal mechanism of coronary blood flow regulation at rest. Furthermore, adenosine is dispensable for the coronary vasodilation during exercise.
Adenosine triphosphate is a potent coronary vasodilator progressively released from red blood capsules via endothelial P2Y receptors when oxygen partial pressure decays [597].
In humans, the K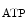 channel contributes to coronary vasodilation during increased myocardial metabolic activity. The K
channel contributes to coronary vasodilation during increased myocardial metabolic activity. The K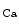 channel that abounds in coronary smooth myocytes (prominently BK
channel that abounds in coronary smooth myocytes (prominently BK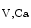 ) also hyperpolarizes the plasma membrane, thereby closing voltage-gated calcium channels to produce vasodilation. Membrane depolarization and elevated cytosolic Ca
) also hyperpolarizes the plasma membrane, thereby closing voltage-gated calcium channels to produce vasodilation. Membrane depolarization and elevated cytosolic Ca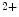 concentration activators of BK
concentration activators of BK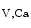 channels, thereby providing a negative feedback that counteracts vasoconstriction. Various protein kinases phosphorylate the K
channels, thereby providing a negative feedback that counteracts vasoconstriction. Various protein kinases phosphorylate the K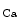 channel, either activating (PKA and PKG) or inhibiting (PKC) the channels. The K
channel, either activating (PKA and PKG) or inhibiting (PKC) the channels. The K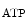 and K
and K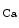 can cooperate to regulate coronary resistive vessel tone during exercise.
can cooperate to regulate coronary resistive vessel tone during exercise.
channel contributes to coronary vasodilation during increased myocardial metabolic activity. The K channel that abounds in coronary smooth myocytes (prominently BK) also hyperpolarizes the plasma membrane, thereby closing voltage-gated calcium channels to produce vasodilation. Membrane depolarization and elevated cytosolic Ca concentration activators of BK channels, thereby providing a negative feedback that counteracts vasoconstriction. Various protein kinases phosphorylate the K channel, either activating (PKA and PKG) or inhibiting (PKC) the channels. The K and K can cooperate to regulate coronary resistive vessel tone during exercise.Voltage-gated outwardly rectifying K channels also lodge on the vascular smooth myocyte. Upon depolarization, they open and thus oppose vasoconstriction. In addition, they are sensitive to β-adrenoceptor stimulation, cAMP-mediated vasodilatory response, and redox signaling (ROS).
channels also lodge on the vascular smooth myocyte. Upon depolarization, they open and thus oppose vasoconstriction. In addition, they are sensitive to β-adrenoceptor stimulation, cAMP-mediated vasodilatory response, and redox signaling (ROS).
channels also lodge on the vascular smooth myocyte. Upon depolarization, they open and thus oppose vasoconstriction. In addition, they are sensitive to β-adrenoceptor stimulation, cAMP-mediated vasodilatory response, and redox signaling (ROS).Impact of the Vasomotor Tone
The total coronary resistance is the sum of structural and vasomotor-associated active components. In the completely vasodilated arterial bed, flow in different perfused cardiac compartments is determined by the luminal area, cumulated arterial length, and the number of in-parallel arteries that supply a given territory. The total length of the arterial circuit irrigating the subendocardium is longer than that of the subepicardial supplier. Most (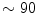 %) of the coronary resistance is linked to small arteries and arterioles.
%) of the coronary resistance is linked to small arteries and arterioles.
%) of the coronary resistance is linked to small arteries and arterioles.During effort, increased cardiac output and vasoconstriction in arterial beds of resting organs raises systolic blood pressure. Vasodilation in arterial beds irrigating exercising muscles buffer this increase, thereby minimizing elevation of diastolic blood pressure. When exercise is maintained, blood pressure often diminishes from its values reached during the early stage of exercise due to a redistribution of blood to the skin for heat dissipation. Cutaneous vasoconstriction at the beginning of exercise is indeed followed by vasodilation for bodily temperature regulation.
Arterial Wall Diameter
The coronary vasomotor tone determines the arterial lumen caliber. Vasoactive substances either bind to their cognate receptors (α- and β-adrenoceptors and muscarinic, adenosine, angiotensin, bradykinin, histamine, endothelin, prostanoids, and serotonin receptors; Vol. 5, Chap. 8. Smooth Myocytes) or cross the plasma membrane to activate their plasma membrane-attached and cortical specific enzymes (e.g., nitric oxide and guanylate cyclase).
Autonomic influences are generally weak. Epicardial vessels primarily have α-adrenoceptors that prime vasoconstriction, whereas intramyocardial and subendocardial vessels predominantly have β2-adrenoceptors that initiate vasodilation. Parasympathetic influences are minor and weakly vasodilatory. During exercise, adrenergic vasoconstriction restrain the increase in coronary blood flow.
Hypoxia causes coronary vasodilation directly and indirectly via adenosine and ATP-sensitive potassium channels.
In addition, the myogenic response to intraluminal pressure changes and metabolic regulation launches fast and slow reaction, respectively.
Most vasoactive messengers operate via the vascular endothelium. Atrial natriuretic peptide, vasoactive intestinal peptide, and calcitonin gene-related peptide cause endothelium-mediated vasodilation, but vasopressin has little effect on the coronary circulation, although it causes vasoconstriction in stressed subjects. Angiotensin-2 is a potent coronary vasoconstrictor. It also releases endothelin, the strongest vasoconstrictory peptide. On the other hand, angiotensin-converting enzyme inactivates bradykinin, a vasodilator.
The vascular endothelium secretes vasorelaxants (nitric oxide, endothelium-derived relaxing factor, prostacyclin, and bradykinin) as well as vasoconstrictors (endothelin-1 and thromboxane-A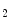 ).
).
).Vasodilation reduces coronary resistance to ensure adequate myocardial perfusion. However, vasodilatory capacity of coronary resistive arteries declines with aging, likely due to reduced availability of nitric oxide, which plays a permissive role in propagating vasomotor signals [608].
In coronary arterioles (caliber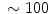 μm) dissected from right atrial appendages of patients, local administration of the endothelium-dependent vasoactive agent bradykinin (100 μmol) elicits vasodilation at local and 2 distant stations (0.5 [station 1] and 1.0 mm [station 2]) from the injection site. The magnitude of vasodilation rises with the duration of stimulus (
μm) dissected from right atrial appendages of patients, local administration of the endothelium-dependent vasoactive agent bradykinin (100 μmol) elicits vasodilation at local and 2 distant stations (0.5 [station 1] and 1.0 mm [station 2]) from the injection site. The magnitude of vasodilation rises with the duration of stimulus (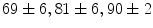 % after 1, 3, and
% after 1, 3, and 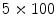 ms, respectively). Bradykinin-induced dilation is substantial at distant sites (
ms, respectively). Bradykinin-induced dilation is substantial at distant sites (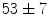 and
and 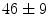 % at stations 1 and 2) [608]. The distant vasodilation, but not the local response, depends on gap junction. Small and intermediate conductance calcium-activated potassium channels (SK
% at stations 1 and 2) [608]. The distant vasodilation, but not the local response, depends on gap junction. Small and intermediate conductance calcium-activated potassium channels (SK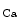 and IK
and IK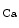 ) are involved in both local and distant sites.
) are involved in both local and distant sites.
μm) dissected from right atrial appendages of patients, local administration of the endothelium-dependent vasoactive agent bradykinin (100 μmol) elicits vasodilation at local and 2 distant stations (0.5 [station 1] and 1.0 mm [station 2]) from the injection site. The magnitude of vasodilation rises with the duration of stimulus ( % after 1, 3, and ms, respectively). Bradykinin-induced dilation is substantial at distant sites ( and % at stations 1 and 2) [608]. The distant vasodilation, but not the local response, depends on gap junction. Small and intermediate conductance calcium-activated potassium channels (SK and IK) are involved in both local and distant sites.The distant vasodilation, but not the local response, is significantly reduced in older (≥64 year-old) patients [608]. Focal application of bradykinin in human coronary arterioles triggers SK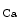 – and IK
– and IK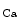 -mediated hyperpolarization that spreads through gap junctions and subsequently remote vasodilation. On the other hand, in younger (
-mediated hyperpolarization that spreads through gap junctions and subsequently remote vasodilation. On the other hand, in younger (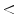 64 year-old) individuals, nitric oxide does not affect local response, but markedly reduces distant vasodilation.
64 year-old) individuals, nitric oxide does not affect local response, but markedly reduces distant vasodilation.
– and IK-mediated hyperpolarization that spreads through gap junctions and subsequently remote vasodilation. On the other hand, in younger (64 year-old) individuals, nitric oxide does not affect local response, but markedly reduces distant vasodilation.During effort, a short-term fast adjustment relies on coronary vasodilation, hence on lowered coronary arterial resistance. Exercise triggers local, nervous, and hormonal regulatory mechanisms to match oxygen demands of the left ventricle during heavy exercise (∼ sixfold increase) by augmenting the coronary blood flow (∼ five times), as hemoglobin concentration and oxygen extraction elevate only modestly [597]. Various types of vasodilators are released (e.g., adenosine, atrial natriuretic peptide, carbon dioxide, lactic acid, and potassium ions).
However, the coronary microvasculature is not maximally dilated, as it retains a vasodilatory reserve during exercise-induced ischemia. In addition, it remains sensitive to vasoconstrictors, such as angiotensin-2, serotonin, thromboxane-A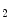 , and vasopressin.
, and vasopressin.
, and vasopressin.Simultaneously, increased sympathetic stimulation and adrenaline secretion from the adrenal medulla cause vasoconstriction of visceral and cutaneous blood vessels as well as vasodilation of blood vessels in skeletal muscles; blood is shunted from the vasculature of the viscera and skin (high resistance) to that of skeletal muscles (low resistance).
Exercise training enables morphometric long-term adaptation of the coronary microvasculature via increased arteriolar densities and/or calibers, in addition to capillary recruitment. Moreover, regular exercise stimulates nitric oxide synthase activity.
Maintenance of α- and β-adrenergic activity despite a lower circulating catecholamine level can result from a heightened adrenoceptor responsiveness.
A significant fraction of coronary resistance is related to small arteries that are not strongly regulated by the metabolic control of the myocardium, but are sensitive to vasodilators [597].
A stenosis in an epicardial coronary artery redistributes the myocardial blood flow during exercise away from the subendocardium to the subepicardium [597].
8.1.4.4 Autoregulation and Flow Reserve
Healthy coronary arteries are characterized by autoregulation to maintain coronary blood flow and match the needs of the myocardium (Vol. 6, Chap. 3. Cardiovascular Physiology). Autoregulation is the intrinsic capacity of small arteries and arterioles to maintain a constant blood flow when the perfusion pressure changes, as they constrict in response to increased intraluminal pressure and conversely. In the physiological range, blood flow is nearly independent of the perfusion pressure. The level of the plateau of the autoregulation curve is related to the metabolic state of the heart. Blood flow adjusts to the myocardial metabolism.
Myogenic, Metabolic, and Endothelial Control
The diameter of small resistive arteries and arterioles is locally regulated by hemodynamic stress as well as by chemicals such as metabolites and gases.
Dysregulated myogenic response can cause local ischemia or vasogenic edema. Increased myogenic activity can engender an elevated peripheral resistance and systemic blood pressure.
Changes in resistive arteriole caliber can be adjusted in few seconds. Changes in microvessel diameter is related to three integrated and coordinated autoregulatory mechanisms: myogenic, metabolic, and flow-dependent endothelial control.
The endothelial control and myogenic response are activated first (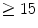 s), and then, if the myocardial contractility and metabolism rise, the metabolic regulation follows.
s), and then, if the myocardial contractility and metabolism rise, the metabolic regulation follows.
s), and then, if the myocardial contractility and metabolism rise, the metabolic regulation follows.Myogenic Control
The myogenic mechanism associated with the vascular smooth myocyte primes vasoconstriction in response to increased stretch of the vascular smooth myocytes and conversely. It is initiated by a stretch-induced depolarization. It involves stretch-sensitive integrins and other types of receptors as well as mechanosensitive ion channels. The depolarization of the plasma membrane activates Ca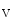 1.2b channel, thereby allowing Ca
1.2b channel, thereby allowing Ca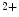 entry.
entry.
1.2b channel, thereby allowing Ca entry.The vascular transmural pressure induces parietal longitudinal and circumferential stresses that is maintained quasi-invariant by the myogenic response. Once the intraluminal pressure increases, the diameter initially rises passively; subsequent smooth muscle contraction causes a diameter reduction and flow resistance elevation, thereby keeping the flow rate and wall stress nearly constant.
When the cytosolic calcium level declines, the myogenic response is maintained by calcium sensitization and actin polymerization via the Rho–PKC pathway. In addition, the PI3K–PKB pathway contributes to the regulation of myogenic tone via activation of the Ca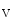 1.2b channel [609].
1.2b channel [609].
1.2b channel [609].Metabolic Control
The metabolic control is driven by metabolites such as adenosine and carbon dioxide, in addition to H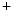 ions, oxygen, and K
ions, oxygen, and K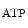 channel. Metabolic autoregulation relies on mutual interference between vascular cells and cardiomyocytes.
channel. Metabolic autoregulation relies on mutual interference between vascular cells and cardiomyocytes.
ions, oxygen, and K channel. Metabolic autoregulation relies on mutual interference between vascular cells and cardiomyocytes.Endothelial Control
The endothelial control results from mechanotransduction, that is, sensing of mechanical forces applied to the vascular wall and its translation into chemical signals that are transmitted from endotheliocytes to adjoining smooth myocytes. Mediators include nitric oxide and endothelium-derived hyperpolarizing factor, among others.
MicroRNA-Mediated Control
An additional control mechanism of myogenic tone involves microRNAs and the miR-processing endonuclease Dicer. MicroRNAs influence vascular smooth myocyte function and intervene in the regulation of the myogenic tone via the PI3K–PKB axis and calcium influx through the Ca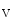 1.2b channel [609]. The loss of myogenic tone in Dicer KO vessels is associated with an increased phosphatase and tensin homolog (PTen) level, abolished stretch-sensitive PKB phosphorylation of myosin light chain, and reduced calcium influx. The PTen phosphatase, a target of several miRNAs, reduce PI3K-mediated PKB phosphorylation by dephosphorylating PIP
1.2b channel [609]. The loss of myogenic tone in Dicer KO vessels is associated with an increased phosphatase and tensin homolog (PTen) level, abolished stretch-sensitive PKB phosphorylation of myosin light chain, and reduced calcium influx. The PTen phosphatase, a target of several miRNAs, reduce PI3K-mediated PKB phosphorylation by dephosphorylating PIP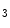 to PIP
to PIP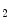 , thereby inhibiting phosphoinositide-dependent kinase PDK1. On the other hand, the PI3K pathway promotes Ca
, thereby inhibiting phosphoinositide-dependent kinase PDK1. On the other hand, the PI3K pathway promotes Ca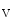 1.2b transfer to the plasma membrane.
1.2b transfer to the plasma membrane.
1.2b channel [609]. The loss of myogenic tone in Dicer KO vessels is associated with an increased phosphatase and tensin homolog (PTen) level, abolished stretch-sensitive PKB phosphorylation of myosin light chain, and reduced calcium influx. The PTen phosphatase, a target of several miRNAs, reduce PI3K-mediated PKB phosphorylation by dephosphorylating PIP to PIP, thereby inhibiting phosphoinositide-dependent kinase PDK1. On the other hand, the PI3K pathway promotes Ca1.2b transfer to the plasma membrane.A combined activity of several miRs may operate in the regulation of the myogenic tone. However, miR26a may be a major element, as it is upregulated by mechanical stretch in airway smooth myocytes [609].
Flow Reserve
Flow reserve at any pressure is the difference between regulated and nonregulated flow. It is influenced by changes in metabolism as well as inflow magnitude, that is, according to whether, the irrigating coronary artery is healthy or stenosed.
The coronary flow reserve (CFR) is the ratio of maximal flow obtained by coronary vasodilation and the resting reference flow. This ratio is reduced in the presence of a stenosis (CFR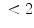 ). Usually, large epicardial coronary arteries contribute slightly to total coronary resistance. When atherosclerosis narrows a large coronary artery with more than 70 % luminal cross-sectional area reduction, hence adding a proximal resistance, autoregulation can preserve basal coronary blood flow, but maximum coronary blood flow is reduced. Consequently, coronary flow reserve is attenuated. The microcirculatory resistance and the maximal dilatory capacity of the microcirculation, together with the severity of the stenosis, determine the resting and the maximal flow. Vasodilation is obtained pharmacologically.
). Usually, large epicardial coronary arteries contribute slightly to total coronary resistance. When atherosclerosis narrows a large coronary artery with more than 70 % luminal cross-sectional area reduction, hence adding a proximal resistance, autoregulation can preserve basal coronary blood flow, but maximum coronary blood flow is reduced. Consequently, coronary flow reserve is attenuated. The microcirculatory resistance and the maximal dilatory capacity of the microcirculation, together with the severity of the stenosis, determine the resting and the maximal flow. Vasodilation is obtained pharmacologically.
). Usually, large epicardial coronary arteries contribute slightly to total coronary resistance. When atherosclerosis narrows a large coronary artery with more than 70 % luminal cross-sectional area reduction, hence adding a proximal resistance, autoregulation can preserve basal coronary blood flow, but maximum coronary blood flow is reduced. Consequently, coronary flow reserve is attenuated. The microcirculatory resistance and the maximal dilatory capacity of the microcirculation, together with the severity of the stenosis, determine the resting and the maximal flow. Vasodilation is obtained pharmacologically.Heterogeneity
Autoregulatory reserve is heterogeneously distributed across the left ventricular wall according to the level of exposure to extravascular compression during systole.
The subendocardium is the most vulnerable region of the cardiac wall to ischemia because of reduced autoregulatory reserve with respect to the subepicardium. The subendocardial coronary arteriolar dynamics exhibit a systolic retrograde flow that is much larger than in the subepicardium [602].
8.1.4.5 Hypoxemia
Ischemia primes a local vasodilator response and recruitment of capillaries within the ischemic region, thereby reducing vascular resistance and augmenting blood flow as well as minimizing the diffusion distance for nutrient transfer. In addition, coronary hypoperfusion can initiate a process within cardiomyocytes that decrease energy demands.
Myocardial ischemia that occurs during exercise in the presence of a coronary arterial stenosis does not cause maximal vasodilation of the coronary resistive vessels so that a substantial vasodilator reserve exists in the terminal arterial bed of the hypoperfused region [597]. This reserve can be recruited using small arterial (e.g., nitrovasodilators) and arteriolar (e.g., adenosine) dilators, due to a persistent vasomotor tone throughout the coronary vasculature.
Whereas adenosine does not participate in the regulation of coronary flow under physiological conditions, it contrastingly contributes to coronary vasodilation in ischemia. Intracoronary infusion of adenosine in the presence of a critical stenosis increases subepicardial flow; the resulting increase in coronary arterial flow raises the pressure drop across the stenosis, thereby further reducing the poststenotic coronary pressure and hence subendocardial blood flow.
Nitric oxide-dependent vasodilation is altered in patients with atherosclerosis, hyperlipidemia, or hypertension [597]. Endothelial dysfunction of the coronary resistive arteries can render patients more vulnerable to hypoperfusion.
In patients with coronary artery disease, inhibition of cyclooxygenase causes coronary vasoconstriction, hence repressing coronary reactive hyperemia and hypoxic coronary vasodilation [597]. Therefore, vasodilatory prostaglandins are involved in the regulation of the coronary vasomotor tone in chronic ischemia.
The K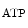 channel that opens and intervenes in the coronary autoregulation is an important mediator of coronary vasodilation during hypoxia and reactive hyperemia.
channel that opens and intervenes in the coronary autoregulation is an important mediator of coronary vasodilation during hypoxia and reactive hyperemia.
channel that opens and intervenes in the coronary autoregulation is an important mediator of coronary vasodilation during hypoxia and reactive hyperemia.Cardiac nerve stimulation constricts resistive vessels in the myocardium irrigated by a stenotic coronary artery via α2-adrenoceptors. Postjunctional α2-adrenoceptor–induced vasoconstriction in ischemic myocardial regions is antagonized by simultaneous stimulation of endothelial α2-adrenoceptor–mediated NO release. α-adrenoceptor–mediated vasoconstriction limits coronary vasodilation in ischemic myocardium, especially in patients with impaired endothelial function.
Moreover, whereas α1- and α2-adrenoceptor stimulation does not affect vessels smaller than 100 μm during normal perfusion, myocardial hypoperfusion is associated with both α1- and α2-adrenoceptor–mediated vasoconstriction in these vessels [597].
Vasoconstriction potentiated by endogenous angiotensin-2 may be partly due to increased noradrenaline release from the sympathetic nerve endings [597]. In addition, hypoperfusion can augment the response of the coronary microvasculature to vasoconstrictory influence such as that of endothelin-1.
Thromboxane-A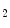 is a product of prostaglandin metabolism liberated during platelet aggregation at sites of intravascular platelet activation and hence thrombus primed by ruptured atherosclerotic plaques. The level of its plasma metabolite thromboxane-B
is a product of prostaglandin metabolism liberated during platelet aggregation at sites of intravascular platelet activation and hence thrombus primed by ruptured atherosclerotic plaques. The level of its plasma metabolite thromboxane-B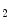 and urinary (2,3)-dinorthromboxane-B
and urinary (2,3)-dinorthromboxane-B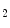 rise in patients with unstable angina [597]. Moreover, thromboxane-A
rise in patients with unstable angina [597]. Moreover, thromboxane-A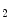 constrict small and large coronary arteries, that is, epicardial and resistive vessels when the coronary artery inflow is normal, whereas adenosine predominantly dilates arterioles. In the presence of a coronary artery stenosis that generates a subendocardial hypoperfusion and ischemic contractile dysfunction in exercising dogs, a TxA
constrict small and large coronary arteries, that is, epicardial and resistive vessels when the coronary artery inflow is normal, whereas adenosine predominantly dilates arterioles. In the presence of a coronary artery stenosis that generates a subendocardial hypoperfusion and ischemic contractile dysfunction in exercising dogs, a TxA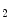 receptor ligand that does not decrease the coronary flow in the normal heart can provoke vasoconstriction that further diminishes myocardial blood flow and aggravates contractile dysfunction. When a stenosis generates a slight but significant decrease in myocardial blood flow with a change from lactate consumption to production, prostacyclin does not oppose the vasoconstriction caused by agonists of thromboxane-A
receptor ligand that does not decrease the coronary flow in the normal heart can provoke vasoconstriction that further diminishes myocardial blood flow and aggravates contractile dysfunction. When a stenosis generates a slight but significant decrease in myocardial blood flow with a change from lactate consumption to production, prostacyclin does not oppose the vasoconstriction caused by agonists of thromboxane-A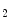 cognate receptors, as the metabolic vasodilation is maximally activated in ischemic myocardium and thus cannot respond to a further decrease in blood flow.
cognate receptors, as the metabolic vasodilation is maximally activated in ischemic myocardium and thus cannot respond to a further decrease in blood flow.
is a product of prostaglandin metabolism liberated during platelet aggregation at sites of intravascular platelet activation and hence thrombus primed by ruptured atherosclerotic plaques. The level of its plasma metabolite thromboxane-B and urinary (2,3)-dinorthromboxane-B rise in patients with unstable angina [597]. Moreover, thromboxane-A constrict small and large coronary arteries, that is, epicardial and resistive vessels when the coronary artery inflow is normal, whereas adenosine predominantly dilates arterioles. In the presence of a coronary artery stenosis that generates a subendocardial hypoperfusion and ischemic contractile dysfunction in exercising dogs, a TxA receptor ligand that does not decrease the coronary flow in the normal heart can provoke vasoconstriction that further diminishes myocardial blood flow and aggravates contractile dysfunction. When a stenosis generates a slight but significant decrease in myocardial blood flow with a change from lactate consumption to production, prostacyclin does not oppose the vasoconstriction caused by agonists of thromboxane-A cognate receptors, as the metabolic vasodilation is maximally activated in ischemic myocardium and thus cannot respond to a further decrease in blood flow.Serotonin constricts epicardial arteries, acting directly on medial smooth myocytes via 5HT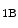 or 5HT
or 5HT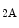 receptors, but dilates coronary resistive vessels likely via 5HT
receptors, but dilates coronary resistive vessels likely via 5HT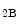 as well as 5HT
as well as 5HT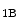 and 5HT
and 5HT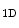 receptors on endotheliocytes, thereby releasing NO [597]. Thus, in epicardial arteries, endothelium-dependent vasodilation engendered by serotonin cannot compensate its direct constrictory action. In the normal heart, dilation of the resistive vessels by serotonin outweighs the effect of constriction of the penetrating arteries. However, in the presence of a coronary artery stenosis that causes arterioles to undergo metabolic vasodilation, vasoconstriction of the penetrating arteries cannot be counterbalanced by additional arteriolar vasodilation.
receptors on endotheliocytes, thereby releasing NO [597]. Thus, in epicardial arteries, endothelium-dependent vasodilation engendered by serotonin cannot compensate its direct constrictory action. In the normal heart, dilation of the resistive vessels by serotonin outweighs the effect of constriction of the penetrating arteries. However, in the presence of a coronary artery stenosis that causes arterioles to undergo metabolic vasodilation, vasoconstriction of the penetrating arteries cannot be counterbalanced by additional arteriolar vasodilation.
or 5HT receptors, but dilates coronary resistive vessels likely via 5HT as well as 5HT and 5HT receptors on endotheliocytes, thereby releasing NO [597]. Thus, in epicardial arteries, endothelium-dependent vasodilation engendered by serotonin cannot compensate its direct constrictory action. In the normal heart, dilation of the resistive vessels by serotonin outweighs the effect of constriction of the penetrating arteries. However, in the presence of a coronary artery stenosis that causes arterioles to undergo metabolic vasodilation, vasoconstriction of the penetrating arteries cannot be counterbalanced by additional arteriolar vasodilation.In summary, the residual coronary vasomotor tone in ischemic myocardium downstream from a coronary stenosis enables vasodilation engendered by adenosine, bradykinin, nitric oxide, and prostanoids, among others, via activation of K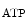 and K
and K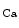 channels. Vasodilation is competed by vasoconstrictory influences of α-adrenoceptor, angiotensin-2, endothelin-1, thromboxane-A
channels. Vasodilation is competed by vasoconstrictory influences of α-adrenoceptor, angiotensin-2, endothelin-1, thromboxane-A , and serotonin that can target small arteries that account for a significant fraction of total coronary resistance, but are not under metabolic control, as well as arterioles under metabolic control.
, and serotonin that can target small arteries that account for a significant fraction of total coronary resistance, but are not under metabolic control, as well as arterioles under metabolic control.
and K channels. Vasodilation is competed by vasoconstrictory influences of α-adrenoceptor, angiotensin-2, endothelin-1, thromboxane-A, and serotonin that can target small arteries that account for a significant fraction of total coronary resistance, but are not under metabolic control, as well as arterioles under metabolic control.8.2 Clinical Scores
The severity of coronary arterial stenoses, that is, the degree of narrowing and number of arterial stenoses, their extent, that is, the proportion of abnormal coronary segments, and pattern, that is, whether plaques are discrete or diffuse, can be quantified using indexes and scores.
The angiographic analysis evaluates CAD severity and extension using the Bogaty [610], Gensini [611], Sullivan [612] scores, among others, as well as collaterals using the Rentrop score [613].
Angiographic scoring systems are strongly correlated with each other and with atherosclerotic plaque burden [614]. However, the involved arteries include according to the group of investigators either all epicardial and branch vessels, all epicardial vessels only, or some epicardial vessels (i.e., no left main coronary artery). Moreover, the severity of lesion comprises varying degrees of stenosis, a single degree of stenosis only; or one degree of stenosis only without recognizing significance criteria. In addition, the functional impact is quantified for the major epicardial and branch vessels, for selected arteries only, or a same significance is assigned to all vessels and their targets.
Apolipoprotein-B is the strongest predictor of both extent and stenosis scores [612]. However, it is more closely related to the extent score, even after correction for age and gender.
8.2.1 Bogaty Score
The severity indexes considered in [610] include: (1) the number of major epicardial vessels with a luminal narrowing 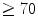 % (maximal number 3), a left main stem stenosis
% (maximal number 3), a left main stem stenosis 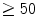 % counting as 2 vessels; (2) the number of narrowings equal or exceeding 50 %, a stenosis length twice the normal lumen diameter being counted as 2 stenoses. A maximum of three stenoses per coronary arterial segment is allowed. Nonocclusive stenoses are classified as concentric, type-1 eccentric, or complex (type-2 eccentric or multiple irregularities).
% counting as 2 vessels; (2) the number of narrowings equal or exceeding 50 %, a stenosis length twice the normal lumen diameter being counted as 2 stenoses. A maximum of three stenoses per coronary arterial segment is allowed. Nonocclusive stenoses are classified as concentric, type-1 eccentric, or complex (type-2 eccentric or multiple irregularities).
% (maximal number 3), a left main stem stenosis % counting as 2 vessels; (2) the number of narrowings equal or exceeding 50 %, a stenosis length twice the normal lumen diameter being counted as 2 stenoses. A maximum of three stenoses per coronary arterial segment is allowed. Nonocclusive stenoses are classified as concentric, type-1 eccentric, or complex (type-2 eccentric or multiple irregularities).The extent score of an arterial segment defined in [610] equals 0 if the segment is angiographically normal; 1 if any abnormality is confined to at most 10 % of the arterial segment length; 2 if the plaque length ranges from 10 to 50 % of the segment length; and 3 if the disease affects more than 50 % of the segment length. When a segment is occluded or suboccluded with altered antegrade flow, a score of 2 (or 3 if any lesion upstream from the occlusion involved more than half the nonoccluded portion of the segment) is arbitrarily assigned, whereas downstream segments are not taken into account. The extent score was the total score of the 15 segments of the coronary arterial tree. A dominant right coronary artery has five segments (proximal, mid, distal, posterior descending artery, and posterior left ventricular). The left main stem corresponds to a single segment. The left anterior descending artery has also five segments (proximal, mid, distal, and two diagonals). The circumflex artery (of a nondominant left coronary artery) has four segments (proximal, distal, obtuse marginal, and another marginal). The extent index was the extent score divided by the number of segments that can be properly visualized by antegrade flow. Therefore, the extent index ranges from 0 (score of 0) to 3 (score 45 divided by 15 segments).
A discrete pattern is supposed to correspond to a maximum of three diseased sites that do not involve more than half the arterial segment length, the rest of the angiogram having a normal appearance [610]. An occlusion that is considered as a discrete pattern in the absence of upstream lesion and when it involves less than half the normal segment length. When 2 segments are diseased, a discrete pattern is defined if the total length of the abnormality is smaller than 25 % of the combined length of the 2 segments. The short-length left main stem can be considered as a discrete pattern even if it is diseased over its entire length. A diffuse pattern is anything exceeding the criteria for a discrete pattern, that is, either more than three diseased sites or a lesion over more than 50 % of the segment length.
The quality of collaterals was qualified from absent-to-poor or good-to-excellent [610].
8.2.2 Gensini Score
The Gensini score considers the geometrical severity of lesions, cumulative effects of multiple obstructions, and significance of jeopardized myocardium [614]. A nonlinear score is assigned to each lesion based on the reduction of luminal diameter. A multiplier is applied to each lesion score based upon its location in the coronary tree depending on the functional significance of the area supplied by that segment. The final Gensini score is the sum of the lesion scores.
8.2.3 Coronary Artery Surgery Study (CASS) Score
The Coronary Artery Surgery Study (CASS) score was developed by Ringqvist and coworkers [615]. Three simple indices encompass: (1) the number of vessels diseased; (2) the number of proximal arterial segments diseased; and (3) a left ventricular wall motion score.
Each of the 3 major epicardial vessels with at least 70 % stenosis is assigned 1 point; stenosis greater than or equal to 50 % in the left main coronary artery is considered a 2-vessel disease and assigned 2 points [614]. The final score is the sum of all points and is analogous to single-, double-, or triple-vessel disease in the coronary tree.
8.2.4 Duke–Jeopardy Score
The Duke–Jeopardy Score was developed and later validated in patients with significant coronary artery disease, but without significant left main coronary stenosis (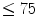 %) [616, 617]. The coronary tree is divided into six segments: the left anterior descending coronary artery, its diagonal branches, septal perforating branches, circumflex coronary artery, obtuse marginal branches, and the posterior descending coronary artery. Each segment distal to at least a 70 % stenosis is assigned 2 points [614]. The maximum number of points is 12.
%) [616, 617]. The coronary tree is divided into six segments: the left anterior descending coronary artery, its diagonal branches, septal perforating branches, circumflex coronary artery, obtuse marginal branches, and the posterior descending coronary artery. Each segment distal to at least a 70 % stenosis is assigned 2 points [614]. The maximum number of points is 12.
%) [616, 617]. The coronary tree is divided into six segments: the left anterior descending coronary artery, its diagonal branches, septal perforating branches, circumflex coronary artery, obtuse marginal branches, and the posterior descending coronary artery. Each segment distal to at least a 70 % stenosis is assigned 2 points [614]. The maximum number of points is 12.8.2.5 Duke Coronary Artery Disease Severity Score
In the scoring system relying on the Duke coronary artery disease severity index [618], the discontinuous score ranges from 0 to 100 with higher weight given to a high number of involved vessels and numerous proximal lesions, as well as severity of left anterior descending coronary artery stenosis [614].
For the least severe CAD (i.e., one-vessel disease), the survival rate is nearly similar whatever the therapy (revascularization [PTCA or CABG] or medical therapy [618]. For intermediate CAD (i.e., two-vessel disease), revascularization increases the survival rates [618]. For less severe two-vessel CAD, PTCA yields a better prognosis than CABG; for the most severe two-vessel CAD (with a critical lesion of the proximal left anterior descending artery), CABG is a better strategy. For the most severe CAD (i.e., three-vessel disease), CABG leads to a better prognosis than PTCA and medical therapy [618].
8.2.6 Friesinger Score
The Friesinger score ranges from 0 to 5 with higher scores given to increased severity and number of luminal stenoses [619, 614].
8.2.7 Jenkins Score
The Jenkins score explores the coronary arterial tree from eight proximal segments [620]. It assigns a score to each segment based on the maximal degree of luminal stenosis. The points for each lesion are summed, thereby reflecting the extent and severity of atherosclerosis in proximal segments of the coronary tree [614].
The concentrations of several circulating lipoproteins (LDL and the combined effect of LDL
and the combined effect of LDL and VLDL
and VLDL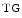 ) are related to the severity of coronary atherosclerosis [620].
) are related to the severity of coronary atherosclerosis [620].
and the combined effect of LDL and VLDL) are related to the severity of coronary atherosclerosis [620].8.2.8 Sullivan Scores
The Sullivan scores represent three distinct scoring techniques [612]. The vessel score that is related to the number of arteries with at least 70 % stenosis ranges from 0 to 3. The left main stenosis is counted as a single-vessel disease [614]. The stenosis score is identical to the Gensini score. The extent score indicates the proportion of atherosclerotic arteries identified by luminal irregularity. A weighting factor takes into account the functional impact. The final score represents the percentage of coronary intimal surface area involved by atherosclerotic plaques.
8.2.9 Agatston Score
The Agatston score is based on the amount of calcium deposits in coronary arterial walls to examine and follow up the progression of coronary artery calcifications (CAC). In the absence of calcifications, the Agatston score equals zero. An Agatston score is computed for each major artery and then summed to get the total score.
The amount of calcifications increases with age. Men develop calcifications about 10–15 years earlier than women [621]. Furthermore, in the majority of asymptomatic men over 55 years and women over 65 years of age, calcifications can be detected. Absolute Agatston scores of less than 10, 11–99, 100–400, and above 400 permit to categorize individuals into groups with minimal, moderate, increased, or extensive amounts of calcification, respectively.
8.2.10 Rentrop Score
The Rentrop score assesses the development of coronary collateralization. The collateral score is based on the opacification quality of collateral arteries after injection of a contrast agent:
Patients can then be classified according to the degree of development of collateral arteries supplying the distal arterial bed downstream from a severe occlusion: collateralization is graded as low (Rentrop score 0–1) or high (Rentrop score 2–3). The collateral score is not correlated with age, smoking habits, hypertension, or total serum, LDL, or HDL cholesterol [622].
0: no collateral vessels;
1: threadlike, poorly opacified collaterals with faint visualization of the distal arterial vasculature;
2: moderately opacified collaterals;
3: large, brightly filled collateral channels with immediate visualization of the entire distal artery.
8.3 Acute Coronary Syndrome
Acute coronary syndrome (ACS) results from atherothrombosis and myocardial ischemia. It includes acute unstable angina, that is, ischemic chest pain at rest or minimal exertion without cardiomyocyte necrosis, and acute myocardial infarction (AMI), that is, ischemic chest pain at rest with cardiomyocyte necrosis, which is subdivided into ST-segment elevation (STEMI) and non-STEMI myocardial infarction.
Rare nonatherosclerotic causes of acute coronary syndrome comprise coronary arteritis, trauma, dissection, thromboembolism, congenital anomalies, cocaine abuse, and complications of cardiac catheterization [623].
8.3.1 Atherosclerotic Plaque Rupture and Erosion
Sudden evolution of the culprit atherosclerotic plaque ranges from thrombosis with or without coronary occlusion to sudden luminal narrowing from intraplaque hemorrhage following plaque rupture and erosion.
Plaque hemorrhage can result from either a plaque rupture or fissure or from angiogenesis. The two main histopathological types of vulnerable plaques include the rupture- and erosion-prone lesions [623].
The ruptured plaque (atheroma or thin-cap fibroatheroma) is characterized by [623]: (1) a big plaque size; (2) a large and soft lipid-rich necrotic core covered by a thin fibrous cap disrupted and infiltrated by foamy macrophages;6 (3) an expansive remodeling (extension of the plaque to adjacent segments and moderate luminal obstruction) with angiogenesis, plaque hemorrhage, adventitial inflammation, and a spotty pattern of calcifications.7
A plaque rupture is a gap in the thin fibrous cap (thickness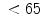 μm) that separates the lipid-rich necrotic core from the arterial lumen. In ruptured plaques, the media is often destroyed. Plaque rupture is the main cause of coronary thrombosis.
μm) that separates the lipid-rich necrotic core from the arterial lumen. In ruptured plaques, the media is often destroyed. Plaque rupture is the main cause of coronary thrombosis.
A plaque fissure is defined as a lateral tear in an eccentric plaque with a small necrotic core. The superficial tear separates a layer of the arterial intima from the underlying fibrous tissue and the hemorrhage extends into the necrotic core; this tract is usually lined by macrophages.
A plaque erosion is related to thrombosis without plaque rupture. Typically, the endothelium is missing at the erosion site. The exposed intima consists predominantly of vascular smooth myocytes and proteoglycans. The plaque morphology shows pathological intimal thickening or a fibroatheroma with an intact media.
A calcified nodule refers to as a disruptive nodular calcification protruding into the lumen that occurs usually in older individuals and in tortuous and heavily calcified arteries.5
Plaque erosion is often associated with [623]: (1) thrombosis, mostly mural; (2) a scarcely calcified plaque; (3) the absence of discontinuation of the fibrous cap; and (4) a sparsely inflamed constrictive remodeling with severe luminal narrowing.
8.3.2 Atherosclerotic Plaque Hemorrhage
Plaque hemorrhage is an important factor of rapid plaque progression. Plaque hemorrhage can originate from plaque rupture. On the other hand, angiogenesis that creates fragile low-pressure neomicrovessels within the plaque derived mainly from vasa vasorum (rarely from the lumen), can be the other source of intraplaque hemorrhage. The latter can contribute to necrotic core expansion and then evolve to a vulnerable plaque. These neomicrovessels lack supporting cells and are leaky, hence allowing local extravasation of plasma proteins and red blood capsules. Angiogenesis and inflammation often coexist at the base of advanced plaques.
The distinction between these two origins of plaque hemorrhage can have therapeutic and prognostic implications.
Plaque fissuring corresponds to the formation of an opening from the arterial lumen into the intima (plaque rupture or fissure) that leads to an intraintimal thrombus containing few red blood capsules and mainly fibrin and platelets.
Pure plaque hemorrhage is defined as the presence of red blood capsules within a plaque originated from small capillaries of the intima coming from the media.
8.3.3 Markers of the Acute Coronary Syndrome
Markers are used in the diagnosis, categorization, and management of patients with acute coronary syndrome. Markers of plaque instability, such as myeloperoxidase, the S100 calcium-binding proteic dimer S100a8–S100a9,8 pregnancy-associated plasma protein-A (PAPa or PAPPa),9 and C-reactive protein, have very lowdiagnostic accuracy.
In myocardial infarction, the cardiac troponins cTnnI and cTnnT are released from necrotic cardiomyocytes as intact proteins and degradation products. Their blood concentrations enable the estimation of cardiomyocyte damage, but with a delayed increase. A cTnn value above the 99th percentile of a normal reference population detected by sensitivity and high-sensitivity assays signs myocardial infarction [624].
Alternative markers of cardiomyocyte damage include the small soluble cardiac fatty acid-binding protein (cFABP) involved in the transfer of long-chain fatty acids into the cardiomyocyte. It is released into the blood circulation upon cardiomyocyte injury, more rapidly than bound cardiac troponins.
Copeptin is the C-terminus of provasopressin. It is secreted with vasopressin from the neurohypophysis. It quantifies the stress level and also the mortality risk in various diseases, especially myocardial infarction very early after symptom onset.
Natriuretic peptides, proadrenomedullin, and growth and differentiation factor GDF15 are powerful predictors of mortality in patients with an acute coronary syndrome.
C-reactive protein is a prognostic marker in acute coronary syndrome without significant differences between patients with and without type-2 diabetes [625].
8.4 Context
Smoking seems to promote thrombosis rather than atherosclerosis [623].
8.4.1 Genetic Background, Atherosclerosis, and Myocardial Infarction
A large proportion of the population has a genetic susceptibility for coronary artery disease, which can ultimately evolve toward myocardial infarction [173]. Identification of myocardial infarction gene loci in human subjects at high risk helps at improving prevention.
Susceptibility genes of coronary artery disease have been identified, such as the APO gene cluster and the Mef2A,10 ALOX5AP,11 LTA4H,12 and Tnfsf2 genes [626].
A region in chromosome 9p21 near the CDKN2A and CDKN2B genes is associated with coronary artery disease in four Caucasian populations [627], as well as myocardial infarction [628]. At chromosomal region 9p21.3, a large antisense noncoding RNA gene (ANRIL) affects the regulation of several other genes. It is expressed in cell types affected by atherosclerosis such as vascular smooth myocytes. In healthy individuals homozygous for the risk allele, RNA expression in blood cells of short ANRIL variants increases (2.2-fold) and that long ANRIL variant decreases (1.2-fold) [173]. The risk allele on chromosome 9p21.3 increases the susceptibility of coronary artery disease, stroke, peripheral arterial disease, as well as aneurysm of the aorta and cerebral vessels.
The SLC22A3-LPAL2-LPA gene cluster corresponds to a strong susceptibility locus for coronary artery disease [629]. A coronary artery disease risk locus is also found on 3q22.3 [630]. The chromosomal site 3q22.3 contains the muscular RAS (MRAS or RRAS3) gene, which is widely distributed, especially in the heart; its product (rRas3) is involved in adhesion signaling, hence cell recruitment.
Genetic polymorphism of innate immune genes that encode Toll-like receptors, nucleotide-binding oligomerization domain-like receptors (NLR), and related signaltransduction molecules, such as interleukin-1 receptor-associated kinase IRAK4, is associated with risk of infections, asthma, and atherosclerosis. Signaling from Toll-like receptors is related to the development of several diseases [631].
People with D299G (Asp299Gly) polymorphism associated with TLR413 have lower concentrations of circulating inflammatory cytokines (IL6) and fibrinogen, among others, and a reduced risk for atherosclerosis. The 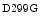 TLR4 polymorphism is also correlated with a decreased risk of bronchoreactivity in patients who inhale lipopolysaccharides in house dust; asthmatic subjects have an increased severity of atopy.14
TLR4 polymorphism is also correlated with a decreased risk of bronchoreactivity in patients who inhale lipopolysaccharides in house dust; asthmatic subjects have an increased severity of atopy.14
TLR4 polymorphism is also correlated with a decreased risk of bronchoreactivity in patients who inhale lipopolysaccharides in house dust; asthmatic subjects have an increased severity of atopy.14 Variants of the ALOX5AP gene are risk factors of myocardial infarction, characterized by higher production in leukotriene-B [633].15 Leukotriene-B
[633].15 Leukotriene-B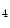 , which is synthesized from leukotriene-A
, which is synthesized from leukotriene-A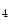 , activates monocytes that migrate across the arterial endothelium and differentiate into macrophages, which become foam cells.
, activates monocytes that migrate across the arterial endothelium and differentiate into macrophages, which become foam cells.
[633].15 Leukotriene-B, which is synthesized from leukotriene-A, activates monocytes that migrate across the arterial endothelium and differentiate into macrophages, which become foam cells.The Tnfsf4 gene is a marker of the risk of myocardial infarction in humans [634]. The myocardium infarction risk rises in coffee consumers with the CYP1A2-1F allele. Susceptibility to stroke is mapped to chromosome 5q12, in particular to the genes encoding phosphodiesterase-4D [635]. Variants in vesicle-associated membrane protein VAMP816 and hnRPUL117 are associated with early-onset myocardial infarction [636].
In addition, the Psrc1, Mia3, and Smad3 genes, which encode the cell-growth regulators Pro/Ser-rich coiled-coil protein-1,18 melanoma inhibitory activity protein-3,19 and SMAD3,20 are significantly associated with myocardial infarction risk.
Several sequence variants that affect eosinophil blood counts suggest association between blood eosinophil number and asthma or myocardial infarction [639]. Asthma (IL1RL1, WDR36, IL33, and MYB) and one myocardial infarction (SH2B3) susceptibility loci have actually been identified.
8.4.2 Inflammation
A local or systemic chronic inflammation arises when antigens are constantly replenished, as in atherosclerosis that persistently harbors antigens. The atherosclerotic lesion is infiltrated by cellular effectors of immunity, such as T and B cells as well as macrophages and dendritic and plasma cells. Inflammation involved in the progression of atherosclerosis is mainly related to macrophages and T lymphocytes (Sect. 1.5).
8.4.2.1 Lymphoid Neogenesis
To eradicate pathogens, the immune system optimizes the likelihood of encounters between antigen-specific T and B lymphocytes of the adaptive immunity with antigen-presenting cells of the innate immunity in secondary lymphoid organs (SLO), such as the spleen and lymph nodes.
During SLO organogenesis, CD3-, CD4+, NR1f3-2+, IL7R+, PTPRc+ lymphoid tissue inducer (LTI) cells that produce TNFSF2 and TNFSF3 interact with stromal lymphoid tissue organizer (LTO) cells and trigger a TNFRSF3-mediated cascade that increases production of adhesion molecules (e.g., ICAM1, MAdCAM1, and VCAM1) and chemokines (CCL19 and CCL21, which attract T and dendritic cells, and CXCL13, which recruits B cells) [640].
The LTI–LTO interaction relies on several ligand–receptor complexes, such as α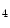 β
β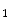 Itg–VCAM1, TNFSF1–TNFR1, and membrane-bound TNFSF2–TNFSFR3 and TNFSF3–TNFSFR3 couples. The receptors TNFR1 (TNFRSF1a), TNFR2 (TNFRSF1b), andTNFSFR3 stimulate the NFκ B pathway, involving the translocation of the P50–RelA and P52–RelB heterodimers to the nucleus, respectively. In addition, production of certain growth factors enables lymphangiogenesis.
Itg–VCAM1, TNFSF1–TNFR1, and membrane-bound TNFSF2–TNFSFR3 and TNFSF3–TNFSFR3 couples. The receptors TNFR1 (TNFRSF1a), TNFR2 (TNFRSF1b), andTNFSFR3 stimulate the NFκ B pathway, involving the translocation of the P50–RelA and P52–RelB heterodimers to the nucleus, respectively. In addition, production of certain growth factors enables lymphangiogenesis.
βItg–VCAM1, TNFSF1–TNFR1, and membrane-bound TNFSF2–TNFSFR3 and TNFSF3–TNFSFR3 couples. The receptors TNFR1 (TNFRSF1a), TNFR2 (TNFRSF1b), andTNFSFR3 stimulate the NFκ B pathway, involving the translocation of the P50–RelA and P52–RelB heterodimers to the nucleus, respectively. In addition, production of certain growth factors enables lymphangiogenesis. Lymphoid neogenesis, or tertiary lymphoid organ (TLO) formation, refers to as the generation of B-cell follicles surrounded by T-cell regions that are anatomically and functionally similar to secondary lymphoid organs, using many of the pathways involved in secondary lymphoid organogenesis occurring before birth [640]. Tertiary lymphoid organs can be built within any nonlymphoid tissues subjected to chronic inflammation. They support adaptive immunity induction and maturation.
Lymphoid aggregates that localize all along the aorta are TLOs because they are composed of B-cell follicles surrounded by T cells, a prototypical organization of ectopic germinal centers [641]. They contain two subsets of B cells with presumably different maturation states as well as blood and lymph vessels and fibroblastic reticular cell (FRC)-like cells. They are polarized toward the media. Lymphangiogenesis and angiogenesis associated with the formation of adventitial TLOs may be due to intramural effectors; vSMCs can trigger intramural angiogenesis, as it produces VEGFa; macrophages can induce the formation of lymphatic vessels [641].
During lymphoid neogenesis in the adventitia of atherosclerotic abdominal aorta, medial smooth myocytes underlying the lesion serve as lymphoid tissue organizers. Similarly to LTO cells in SLOs, medial smooth myocytes may be activated via TNFRSF3, thereupon expressing chemokines (e.g., CCL19–CCL21, CXCL13, and CXCL16) and attracting B and T lymphocytes and dendritic cells to the adventitia [641].
Similarly to LTI cells, M1 macrophages that express high levels of TNFSF1 and membrane-bound TNFSF2 stimulate medial smooth myocytes, which then secrete chemokines (CCL19–CCL20 and CXCL16), thereby supporting the development of adventitial TLOs in arterial walls with advanced atherosclerotic plaques [641]. These macrophages liberate TNFSF1 and TNFSF2, the latter existing in membranebound and secreted forms. Secreted TNFSF2 binds to both TNFR1 (TNFRSF1a) and TNFR2 (TNFRSF1b) with high affinity. On the other hand, the transmembrane TNFR2–TNFR3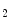 heterotrimer selectively binds to TNFSF3 with high affinity, signaling from which is dispensable for the action of M1 macrophages [641].
heterotrimer selectively binds to TNFSF3 with high affinity, signaling from which is dispensable for the action of M1 macrophages [641].
heterotrimer selectively binds to TNFSF3 with high affinity, signaling from which is dispensable for the action of M1 macrophages [641].Neutralization of TNFSF1 reduces germinal center B cells and adventitial TLOs. Macrophages are major producers of TNFSF1, but B lymphocytes also manufacture TNFSF1 that takes membrane-bound and soluble forms and T lymphocytes may contributes to a complementary TNFSF1 signal [640].
8.4.2.2 Macrophage Populations
ProinflammatoryM1macrophages are antagonized byM2macrophages that dampen inflammation and promote tissue repair. A third type of macrophage, hemoglobin-stimulated macrophage (M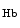 ) is observed at site of hemorrhage or angiogenesis in human atherosclerotic lesions [623].
) is observed at site of hemorrhage or angiogenesis in human atherosclerotic lesions [623].
) is observed at site of hemorrhage or angiogenesis in human atherosclerotic lesions [623].The most common macrophages in the atherosclerotic plaques are foamy M1 macrophages activated by Infγ and T -type cytokines. M2 macrophages are activated by T
-type cytokines. M2 macrophages are activated by T -type cytokines (i.e., IL4 and IL13). CD163+, CD206+ M
-type cytokines (i.e., IL4 and IL13). CD163+, CD206+ M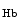 macrophages are devoid of neutral lipids (unlike foamy macrophages) [623]. They express the scavenger receptor HbScaR (CD163; but not ScaRb3), abundantly ATP-binding cassette transporters, and mannose receptor (CD206 or CLec13d). They only slightly produced inducible nitric oxide synthase (NOS2). Adecrease in intracellular iron level may protect from lipid accumulation, partly by reducing intracellular iron-driven generation of reactive oxygen species such as hydroxyl radical upon upregulation of ferroportin expression.
macrophages are devoid of neutral lipids (unlike foamy macrophages) [623]. They express the scavenger receptor HbScaR (CD163; but not ScaRb3), abundantly ATP-binding cassette transporters, and mannose receptor (CD206 or CLec13d). They only slightly produced inducible nitric oxide synthase (NOS2). Adecrease in intracellular iron level may protect from lipid accumulation, partly by reducing intracellular iron-driven generation of reactive oxygen species such as hydroxyl radical upon upregulation of ferroportin expression.
-type cytokines. M2 macrophages are activated by T-type cytokines (i.e., IL4 and IL13). CD163+, CD206+ M macrophages are devoid of neutral lipids (unlike foamy macrophages) [623]. They express the scavenger receptor HbScaR (CD163; but not ScaRb3), abundantly ATP-binding cassette transporters, and mannose receptor (CD206 or CLec13d). They only slightly produced inducible nitric oxide synthase (NOS2). Adecrease in intracellular iron level may protect from lipid accumulation, partly by reducing intracellular iron-driven generation of reactive oxygen species such as hydroxyl radical upon upregulation of ferroportin expression.8.4.2.3 Myeloperoxidase-Positive Cells
The circulating inflammatory marker myeloperoxidase (MPo) is higher in patients with OCT-defined plaque erosion than rupture [623]. The density of MPo+ cells is higher in thrombi caused by eroded plaques than in ruptured plaques in fatal coronary thrombosis.
8.4.3 Oxidative Stress and PKB Kinase
Human mortality results mainly from perturbed lipid metabolism and chronic inflammation associated with atherothrombosis and myocardial infarction. High levels of reactive oxygen species are generated in the heart during ischemia, as oxygen concentration does not immediately fall to zero.
The murine model that recapitulates atherothrombosis leading to death from myocardial infarction is the mouse lacking both the high-density lipoprotein receptor scavenger receptor ScaRb1 and apolipoprotein-E (ApoE , ScaRb1
, ScaRb1 double-knockout mouse). The ScaRb1 receptor launches the main endothelial protective axis especially in hyperlipidemia, as it triggers the PKB–NOS3 pathway, which precludes NFκ B activation induced by VCAM1 on endotheliocytes [642].
double-knockout mouse). The ScaRb1 receptor launches the main endothelial protective axis especially in hyperlipidemia, as it triggers the PKB–NOS3 pathway, which precludes NFκ B activation induced by VCAM1 on endotheliocytes [642].
, ScaRb1 double-knockout mouse). The ScaRb1 receptor launches the main endothelial protective axis especially in hyperlipidemia, as it triggers the PKB–NOS3 pathway, which precludes NFκ B activation induced by VCAM1 on endotheliocytes [642]. The major cardiovascular PKB isoform is PKB1 that represents 50 % of the PKB activity in the heart and 70 % in endotheliocytes [642]. It is also the main isoform in other cells involved in atherothrombosis, such as vascular smooth myocytes, monocytes, and platelets.
The PKB1 subtype that is protective in the endothelium in atherosclerosis, as it supports cell survival, can have detrimental effects when it is excessively stimulated.
In particular, it is activated by oxidized low-density lipoprotein uptake. It is also involved in proinflammatory signaling in smooth myocytes and macrophages. In patients, PKB is overactivated in atherosclerotic plaques, after myocardial infarction, and in heart failure.
In ApoE mice subjected to a Western-type diet, PKB1 deficiency that disrupts the protective NOS3 signaling increases endothelial damage and apoptosis and exacerbates atherosclerotic lesion development. Furthermore, endotheliocyte-specific deletion of the FoxO transcription factor that is degraded upon PKB activation, is atheroprotective. Knockout of three FOXO genes in endotheliocytes attenuates PKB activity. On the other hand, inhibition by rapamycin of the TOR kinase that acts both upstream and downstream of PKB, increases lifespan. In ApoE
mice subjected to a Western-type diet, PKB1 deficiency that disrupts the protective NOS3 signaling increases endothelial damage and apoptosis and exacerbates atherosclerotic lesion development. Furthermore, endotheliocyte-specific deletion of the FoxO transcription factor that is degraded upon PKB activation, is atheroprotective. Knockout of three FOXO genes in endotheliocytes attenuates PKB activity. On the other hand, inhibition by rapamycin of the TOR kinase that acts both upstream and downstream of PKB, increases lifespan. In ApoE , ScaRb1
, ScaRb1 mice characterized by diet-independent changes in lipid profiles with high blood concentrations of cholesterol due to blocked cholesterol accumulation and processing in the liver, the PKB1 level is correlated with cholesterol accumulation in macrophages during atherosclerosis and cardiac dysfunction, hypertrophy, and fibrosis, as well as increased infarct region [642]. It is linked to inflammation, oxidative stress, accumulation of oxidized lipids, and an increased level of ScaRb3, a sensor of oxidative stress that binds to oxLDLs, hence promoting foam cell formation. These events create a positive feedback loop that exacerbate oxidative stress effects. On the other hand, PKB deficiency reduces ROS generation and lipid oxidation, binding, and accumulation due to low ScaRb3 density on macrophages.
mice characterized by diet-independent changes in lipid profiles with high blood concentrations of cholesterol due to blocked cholesterol accumulation and processing in the liver, the PKB1 level is correlated with cholesterol accumulation in macrophages during atherosclerosis and cardiac dysfunction, hypertrophy, and fibrosis, as well as increased infarct region [642]. It is linked to inflammation, oxidative stress, accumulation of oxidized lipids, and an increased level of ScaRb3, a sensor of oxidative stress that binds to oxLDLs, hence promoting foam cell formation. These events create a positive feedback loop that exacerbate oxidative stress effects. On the other hand, PKB deficiency reduces ROS generation and lipid oxidation, binding, and accumulation due to low ScaRb3 density on macrophages.
mice subjected to a Western-type diet, PKB1 deficiency that disrupts the protective NOS3 signaling increases endothelial damage and apoptosis and exacerbates atherosclerotic lesion development. Furthermore, endotheliocyte-specific deletion of the FoxO transcription factor that is degraded upon PKB activation, is atheroprotective. Knockout of three FOXO genes in endotheliocytes attenuates PKB activity. On the other hand, inhibition by rapamycin of the TOR kinase that acts both upstream and downstream of PKB, increases lifespan. In ApoE, ScaRb1 mice characterized by diet-independent changes in lipid profiles with high blood concentrations of cholesterol due to blocked cholesterol accumulation and processing in the liver, the PKB1 level is correlated with cholesterol accumulation in macrophages during atherosclerosis and cardiac dysfunction, hypertrophy, and fibrosis, as well as increased infarct region [642]. It is linked to inflammation, oxidative stress, accumulation of oxidized lipids, and an increased level of ScaRb3, a sensor of oxidative stress that binds to oxLDLs, hence promoting foam cell formation. These events create a positive feedback loop that exacerbate oxidative stress effects. On the other hand, PKB deficiency reduces ROS generation and lipid oxidation, binding, and accumulation due to low ScaRb3 density on macrophages.8.4.4 Impaired Mitochondrial Structure and Function
Mitochondria are sensitive to ischemic insult and trigger apoptosis. Mitochondrial morphology dynamics affect the outcome of cardiac ischemia and ischemia–reperfusion injury.
8.4.4.1 Mitochondrial Morphology Dynamics
Mitochondria constantly undergo fusion or fission and present either a reticular elongated or punctate fragmented morphotype, respectively, thereby allowing elimination of dysfunctional mitochondria in the latter case.
Mitochondrial fusion involves the mitochondrial guanosine triphosphatases (GTPase) dynamin-like Optic atrophy protein OpA1 and transmembrane mitofusins Mfn1 and Mfn2. On the other hand, mitochondrial fission relies on the mitochondrial outer membrane dynamics protein fission-1 homolog (Fis1) and dynamin-related protein DRP1 (or dynamin 1-like protein Dnm1L).
The profission protein DRP1 shuttles between the cytosol and mitochondria. Dephosphorylation of DRP1 (Ser637) prevents cytosolic sequestration of DRP1, thereby launching mitochondrial fission. In addition, BCL2-binding component BBC3 (or P53 upregulated modulator of apoptosis [PUMA]), an activator of the mitochondrial apoptotic and cell death pathway, as it sequesters BCL2 and BCLxL and activates proapoptotic BAX and/or BAK, supports DRP1 activity [643].
The dynamin-related GTPase DRP1 mediates cardiac cell death during ischemic damage. On the other hand, the antiapoptotic and proproliferative Ser/Thr kinase PIM1 is an effector of PKB-mediated cardioprotection that preserves mitochondrial integrity and prevents initiation of the intrinsic mitochondrial apoptotic pathway. The kinase PIM1 phosphorylates DRP1 (Ser637), thereby precluding DRP1 localization to mitochondria and preserving reticular mitochondrial morphology in response to ischemic stress [643]. The cardioprotective PIM1 thus prevents mitochondrial fission and maintains the mitochondrial structure during cardiac ischemia, thereby promoting regeneration of the myocardium after myocardial infarction.
8.4.4.2 Oxidative Phosphorylation
The maximal oxidative phosphorylation capacity in ischemic myofibers lowers compared with nonischemic myocardium [644]. Diminished mitochondrial oxidative phosphorylation capacity and excessive production of reactive oxygen species engendered by ischemia alter the mitochondrial function in the myocardium. Chronic myocardial ischemia damages mitochondria, hence not only compromising ATP synthesis, but also producing ROS by the electron transport chain. Excessive ROS production inflicts additional damage of mitochondrial membrane constituents (lipids and proteins) and DNA, thereby opening of mitochondrial permeability transition pores that engenders mitochondrial depolarization, cytochrome-C loss, and apoptosis, ultimately leading to cardiomyopathy. Moreover, the antioxidant level is attenuated.
Blockade of  complex-I or -III decreases ROS production during episodes of ischemia and helps protect mitochondria against ischemic damage [644].
complex-I or -III decreases ROS production during episodes of ischemia and helps protect mitochondria against ischemic damage [644].  complex-II may also manufacture ROS as in skeletal myocytes.
complex-II may also manufacture ROS as in skeletal myocytes.
complex-I or -III decreases ROS production during episodes of ischemia and helps protect mitochondria against ischemic damage [644]. complex-II may also manufacture ROS as in skeletal myocytes.Alterations in the kinetic properties of cytochrome-C oxidase ( complex-IV) can impair electron handling and generate ROS in excess. However, its capacity in the ischemic and nonischemic regions under both hyperoxic and hypoxic conditions does not vary markedly [644]. Nevertheless, the activity of cytochrome-C oxidase subunit-4 is regulated allosterically by the energetic level within the cell. In addition, the composition of cytochrome-C oxidase subunits change with hypoxia. Yet, affected cytochrome-C oxidase composition is likely a primary element in mitochondrial dysfunction in CIHD conditions.
complex-IV) can impair electron handling and generate ROS in excess. However, its capacity in the ischemic and nonischemic regions under both hyperoxic and hypoxic conditions does not vary markedly [644]. Nevertheless, the activity of cytochrome-C oxidase subunit-4 is regulated allosterically by the energetic level within the cell. In addition, the composition of cytochrome-C oxidase subunits change with hypoxia. Yet, affected cytochrome-C oxidase composition is likely a primary element in mitochondrial dysfunction in CIHD conditions.
complex-IV) can impair electron handling and generate ROS in excess. However, its capacity in the ischemic and nonischemic regions under both hyperoxic and hypoxic conditions does not vary markedly [644]. Nevertheless, the activity of cytochrome-C oxidase subunit-4 is regulated allosterically by the energetic level within the cell. In addition, the composition of cytochrome-C oxidase subunits change with hypoxia. Yet, affected cytochrome-C oxidase composition is likely a primary element in mitochondrial dysfunction in CIHD conditions.On the other hand,  complex-II oxidative phosphorylation capacity diminishes in chronically ischemic left ventricular myocardium in humans [644].
complex-II oxidative phosphorylation capacity diminishes in chronically ischemic left ventricular myocardium in humans [644].
complex-II oxidative phosphorylation capacity diminishes in chronically ischemic left ventricular myocardium in humans [644].8.4.5 Proteasomal Activity
The ubiquitin–proteasome route of protein degradation is the major nonlysosomal proteolytic system that regulates protein turnover, controls the cell cycle, antigen presentation, and inflammation.
The imbalance between myocardial oxygen demand and supply during ischemia engenders a progressive depletion of the cellular ATP content (normal cardiac ATP concentration 4–6 mmol decays to 35, 16, 9, and 7 % of normal level after 15, 30, 40, and 60 min of ischemia, respectively [645]).
During protein ubiquitination catalyzed by tripartite motif-containing ubiquitin–protein ligase TRIM63,21 double minute-2, atrogin-1, RING finger protein RNF41,22 STIP1 homology and U-box–containing ubiquitin–protein ligase STUB1,23 ATP activates the C-terminal glycine of ubiquitin via the generation of an adenylate intermediate by an E1 enzyme, followed by the formation of an E1–ubiquitin thiolester and release of AMP and inorganic pyrophosphate.
The 26S proteasome is constituted of the multimeric 20S core proteasome that is singly or doubly capped at its ends by a 19S regulator complex. The proteasomal core particle is composed of four stacked rings, each consisting of seven distinct α and β subunits (α1–α7 and β1–β7). The activity of the β1, β2-, and β5 proteolytic subunits are referred to as caspase-like, trypsin-like, and chymotrypsin-like, respectively [645]). The 19S regulator is made up at least of 17 subunits, including 6 ATPases associated with different cellular activities (Rpt1–Rpt6), 3 non-ATPase subunits (Rpn1, 2, and 10) that interact with the 20S proteasome, and a lid of 8 nonATPase subunits. It confers dependency on ATP and Mg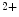 and specificity for ubiquitinated substrate [645]).
and specificity for ubiquitinated substrate [645]).
and specificity for ubiquitinated substrate [645]). The ATP fuel is required for the 26S proteasome assembly and stability. Whereas ADP, AMP, adenosine, and inorganic pyrophosphate cannot substitute for the effect of ATP on the 26S proteasomal peptidase, the 26S proteasome has a broad specificity for nucleotides with a preference for ATP with respect to nonadenine nucleotides (ATP>CTP≫GTP>UTP) in cardiomyocytes [645]).
The 26S proteasome is split into the 19S regulator and 20S core particle upon ATP depletion. Upon readdition of ATP, the 19S regulator and 20S particle reform the 26S proteasome [645]). In addition, ATP binding and hydrolysis also regulate degradation of ubiquitinated proteins by the 26S proteasome.
Depletion of the myocardial ATP content disassembles the 26S proteasome and activates its ATP-dependent peptidase activity and, hence, abnormal degradation of ubiquitin–protein conjugates. The proteasomal peptidase activity is likely attributable to 26S proteasomes that remain intact at very lowATPconcentrations [645]). Elevated proteasomal degradation of proteic substrates, such as AMPK, GRK2, RyR2, Cx43, and NKκ B, and possibly of myofibrillar proteins, contributes to ischemic myocardial injury [645]).
On the other hand, the proteasomal activity lowers in the postischemic heart. Ischemic preconditioning that reduces the rate of ATP depletion during subsequent sustained ischemia attenuates the increase of the myocardial proteasomal activity and preserves the 26S proteasomal function in the postischemic heart via diminished oxidative damage of the 19S regulator or interactions with protein kinases PKA and PKC [645]). However, ischemic preconditioning causes a net inhibition of the cardiac 26S proteasome during ischemia.
Proteasome inhibitors during cardiac ischemia and reperfusion can have cardioprotective effects when administered before or during ischemia. In addition, proteasome inhibitors may be beneficial, as they have antiinflammatory and immunosuppressive actions that attenuate leukocyte-mediated myocardial reperfusion injury [645]). However, proteasome inhibitors have side effects and toxicities.
8.4.6 Influence of Obesity
The coronary perivascular adipose tissue influences the coronary vasomotor tone via secreted factors, potentiating contraction of coronary vascular smooth myocytes, at least in obese swine with respect to lean swine [646]. The coronary adipose tissue (and mesenteric, but not subcutaneous adipose tissue) augments coronary contractions after exposure to KCl (20 mmol/L) as well as to prostaglandin-F2α in proportion to the amount of perivascular adipose tissue in both intact and endothelium-denuded arteries. The coronary perivascular adipose tissue also diminishes H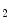 O
O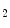 -mediated vasodilation in arteries of lean swine and, to a lesser extent, in arteries of obese swine. Vascular effects of the coronary perivascular adipose tissue vary according to anatomic location [646]. Augmented contractile effects of obese coronary perivascular adipose tissue are related to alterations in the perivascular adipose tissue proteome (e.g., calpastatin that elevates vSMC contraction), Rho–RoCK signaling, and K
-mediated vasodilation in arteries of lean swine and, to a lesser extent, in arteries of obese swine. Vascular effects of the coronary perivascular adipose tissue vary according to anatomic location [646]. Augmented contractile effects of obese coronary perivascular adipose tissue are related to alterations in the perivascular adipose tissue proteome (e.g., calpastatin that elevates vSMC contraction), Rho–RoCK signaling, and K and Ca
and Ca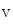 1.2 channels.
1.2 channels.
O-mediated vasodilation in arteries of lean swine and, to a lesser extent, in arteries of obese swine. Vascular effects of the coronary perivascular adipose tissue vary according to anatomic location [646]. Augmented contractile effects of obese coronary perivascular adipose tissue are related to alterations in the perivascular adipose tissue proteome (e.g., calpastatin that elevates vSMC contraction), Rho–RoCK signaling, and K and Ca1.2 channels.8.4.7 Diabetes
Type-2 diabetes is characterized by potent proinflammatory, prooxidant, and prothrombotic stimuli, hence engendering more frequently acute vascular events than nondiabetic patients. In patient populations with a first acute coronary syndrome, diabetic patients exhibit more severe coronary atherosclerosis than nondiabetic patients [647]. Diabetic patients have a better collateral circulation observed by coronary angiography as well as a more strongly calcified plaque and smaller lipid content at the site of the minimal lumen area and more superficial calcified nodules detected by intracoronary optical coherence tomography. Hence, diabetic patients seem to experience their first event at a later stage of coronary atherosclerosis.
8.5 Obstructive Epicardial Coronary Artery Disease
Coronary artery disease results from atherosclerosis (Vol. 8, Chap. 7. Atherosclerosis—Biological Aspects). After a relatively long delay, atherosclerotic plaque forms stenosis that narrows the coronary arterial lumen and hence reduces the myocardial perfusion. Moreover, the atherosclerotic plaque can rupture; the subsequent blood clot can completely block blood flow or, most likely, can be shed by the flowing blood into emboli that prevent blood passage in downstream arterial branches. In both cases, a myocardial infarction occurs. In addition, CAD weakens the myocardium, thereby provoking heart failure and arrhythmias.
8.6 Coronary Microvascular Dysfunction
Coronary microvascular dysfunction (CMVD), or microvascular angina, resulting from structural and/or functional abnormalities can cause myocardial ischemia, thereby mimicking obstructive epicardial coronary artery disease [648]. Increased coronary microvascular resistance can impair myocardial perfusion and hence occasioning angina with ischemic electrocardiographic changes (e.g., ST-segment depression during exercise), but without arteriographic abnormalities. In otherwords, stress-induced ischemic ECG trace changes are associated with a normal coronary angiogram.
Abnormal coronary microvascular perfusion does not necessarily involved uniformly all coronary microvessels of a major coronary branch, but can be scattered in the myocardium. Small intramural prearteriolar coronary arteries can be the site of coronary microvascular dysfunction and microvascular ischemia.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


