and low-density lipoprotein receptor (LDLR)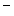 mice can develop atherosclerosis both with and without high-fat feeding, hence their selection as a representative model of human situation. However, lesions that develop over a period of weeks do not mimic accurately those that evolve over decades in humans, in particular fibrous plaques as well as lesions with calcification, ulceration, hemorrhage, and/or thrombosis. Whatever the animal species, some mediators can lack or intervene at a different concentration. Therefore, the process in a given species can vary in comparison with that in another species. For example, cholesteryl ester transferase exists at a much greater level in humans than in mice. Conversely, HDL concentration is higher in mice than humans. In any case, all models are a simplification of the reality.
mice can develop atherosclerosis both with and without high-fat feeding, hence their selection as a representative model of human situation. However, lesions that develop over a period of weeks do not mimic accurately those that evolve over decades in humans, in particular fibrous plaques as well as lesions with calcification, ulceration, hemorrhage, and/or thrombosis. Whatever the animal species, some mediators can lack or intervene at a different concentration. Therefore, the process in a given species can vary in comparison with that in another species. For example, cholesteryl ester transferase exists at a much greater level in humans than in mice. Conversely, HDL concentration is higher in mice than humans. In any case, all models are a simplification of the reality.
2.3 Risk Factors
Age | Point | |
|---|---|---|
Men | Women | |
20–34 | −9 | −7 |
35–39 | −4 | −3 |
40–44 | 0 | 0 |
45–49 | 3 | 3 |
50–54 | 6 | 6 |
55–59 | 8 | 8 |
60–64 | 10 | 10 |
65–69 | 11 | 12 |
70–74 | 12 | 14 |
75–79 | 13 | 16 |
Points | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
20–39 | 40–49 | 50–59 | 60–69 | 70–79 | 20–39 | 40–49 | 50–59 | 60–69 | 70–79 | |
NS | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
S | 8 | 5 | 3 | 1 | 1 | 9 | 7 | 4 | 2 | 1 |
Systolic BP (mmHg) | Men | Women | ||
|---|---|---|---|---|
Untreated | Treated | Untreated | Treated | |
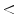 120 120 | 0 | 0 | 0 | 0 |
120–129 | 0 | 1 | 1 | 3 |
130–139 | 1 | 2 | 2 | 4 |
140–159 | 1 | 2 | 3 | 5 |
≥ 160 | 2 | 3 | 4 | 6 |
Points | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
Men | Women | |||||||||
Age | 20–39 | 40–49 | 50–59 | 60–69 | 70–79 | 20–39 | 40–49 | 50–59 | 60–69 | 70–79 |
< 160 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
160–199 | 4 | 3 | 2 | 1 | 0 | 4 | 3 | 2 | 1 | 1 |
200–239 | 7 | 5 | 3 | 1 | 0 | 8 | 6 | 4 | 2 | 1 |
240–279 | 9 | 6 | 4 | 2 | 1 | 11 | 8 | 5 | 3 | 2 |
≥280 | 11 | 8 | 5 | 3 | 1 | 13 | 10 | 7 | 4 | 2 |
2.3.1 Lifestyle
2.3.1.1 Inactivity
2.3.1.2 Diet
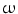 3-fatty acids such as docosahexaenoic acid (DHA) that abound in oily fish (e.g., anchovy, herring, mackerel, and salmon) protect the immune, nervous, and cardiovascular systems. In vascular smooth myocytes, DHA (but not its ethyl ester derivative) directly, rapidly, potently, and reversibly activates large-conductance, voltage- and Ca
3-fatty acids such as docosahexaenoic acid (DHA) that abound in oily fish (e.g., anchovy, herring, mackerel, and salmon) protect the immune, nervous, and cardiovascular systems. In vascular smooth myocytes, DHA (but not its ethyl ester derivative) directly, rapidly, potently, and reversibly activates large-conductance, voltage- and Ca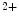 -activated K
-activated K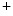 channels (BK or K
channels (BK or K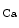 1.1)3 that acts as a vasodilator, thereby lowering blood pressure [113]. The BK channel is activated by intracellular Ca
1.1)3 that acts as a vasodilator, thereby lowering blood pressure [113]. The BK channel is activated by intracellular Ca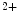 and depolarization. It keeps the membrane hyperpolarized (negative feedback on cellular excitability). This proteic complex operates as a high-affinity receptor for DHA without needing Ca
and depolarization. It keeps the membrane hyperpolarized (negative feedback on cellular excitability). This proteic complex operates as a high-affinity receptor for DHA without needing Ca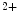 ions. Lipid DHA is released from the plasma membrane by G-protein-activated, Ca
ions. Lipid DHA is released from the plasma membrane by G-protein-activated, Ca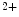 -dependent phospholipase-A2. The concentration required to activate BK channel is about 20 times lower than that needed to stimulate GPR120 involved in anti-inflammatory action of DHA [113].
-dependent phospholipase-A2. The concentration required to activate BK channel is about 20 times lower than that needed to stimulate GPR120 involved in anti-inflammatory action of DHA [113].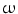 3-fatty acids refer to esterification with ethanol (i.e., ethyl esters) or with glycerol as triglycerides. Various
3-fatty acids refer to esterification with ethanol (i.e., ethyl esters) or with glycerol as triglycerides. Various 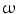 3 and
3 and 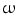 6-fatty acids as well as their ethyl and glycerol ester derivatives have distinct effects on BK channels and blood pressure when acutely applied. In particular, ethyl ester of DHA fails to reduce blood pressure. Oral or parenteral administration of these products thus has different clinical impact. In addition, the physiological response of healthy individuals and patients to various types of fatty acids may differ appreciably.
6-fatty acids as well as their ethyl and glycerol ester derivatives have distinct effects on BK channels and blood pressure when acutely applied. In particular, ethyl ester of DHA fails to reduce blood pressure. Oral or parenteral administration of these products thus has different clinical impact. In addition, the physiological response of healthy individuals and patients to various types of fatty acids may differ appreciably.HDL (mg/dl) | Points | |
|---|---|---|
Men | Women | |
≥60 | − 1 | − 1 |
50–59 | 0 | 0 |
40–49 | 1 | 1 |
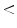 40 40 | 2 | 2 |
flavonols (e.g., kaempferol, myrestin, and quercetin [in onions]);
flavanols (e.g., catechin and epicatechin [in cocoa, red wine, green tea, and apples]);
flavones (e.g., apigenin and luteolin [in peppers]);
isoflavones;
flavanones (e.g., eriodictyol, hesperetin, and naringenin [in oranges]); and
anthocyanidines (e.g., cyanidin, delphinidin, malvedin, and pelargonidin [in blueberries]), which are defined by the chemical residues attached to the basic flavonoid structure.
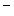 catechin and
catechin and 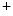 catechin as well as
catechin as well as 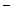 epicatechin and
epicatechin and 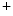 epicatechin) can vary considerably according to the food type.
epicatechin) can vary considerably according to the food type.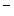 epicatechin (5–250 nmol) represents a minor part of total plasma flavanols (
epicatechin (5–250 nmol) represents a minor part of total plasma flavanols (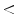 3
3 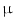 mol; short half-life) [115]. Epicatechin metabolites include methylated, glucuronidated, and sulfated adducts. Monomeric flavanols are further processed in the liver.
mol; short half-life) [115]. Epicatechin metabolites include methylated, glucuronidated, and sulfated adducts. Monomeric flavanols are further processed in the liver.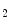 -dependent vasodilation.
-dependent vasodilation. OA]) and other free and esterified fatty acid nitroalkenes are formed at elevated levels when unsaturated fatty acids are ingested together with a source of nitrite [123].
OA]) and other free and esterified fatty acid nitroalkenes are formed at elevated levels when unsaturated fatty acids are ingested together with a source of nitrite [123].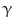 (nuclear receptor NR1c3), the stress sensor and NFE2L2 sequestrator Kelch-like erythroid cell-derived protein with CNC homology (EHC)-associated protein KEAP1,6 and nuclear factor erythroid-derived-like factor NFE2L2-regulated antioxidant response genes, and inhibition of proinflammatory gene expression regulated by NF
(nuclear receptor NR1c3), the stress sensor and NFE2L2 sequestrator Kelch-like erythroid cell-derived protein with CNC homology (EHC)-associated protein KEAP1,6 and nuclear factor erythroid-derived-like factor NFE2L2-regulated antioxidant response genes, and inhibition of proinflammatory gene expression regulated by NF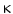 B [123]. Nitrofatty acids also inhibit lipopolysaccharide-induced cytokine expression and induce HO1 expression via activation of NFE2L2-regulated gene expression. Therefore, electrophilic lipid derivatives can control gene transcription that overall engenders an anti-inflammatory response.
B [123]. Nitrofatty acids also inhibit lipopolysaccharide-induced cytokine expression and induce HO1 expression via activation of NFE2L2-regulated gene expression. Therefore, electrophilic lipid derivatives can control gene transcription that overall engenders an anti-inflammatory response.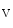 1.2b channel. Nitro-oleic acid activates TRPA1 and TRPV1 channels in sensory neurons involved in neurogenic inflammation and pain induced by noxious chemicals or thermal stimuli, thereby provoking calcium influx, membrane depolarization, and firing. In addition, high concentrations of nitro-oleic acid suppress firing in dorsal root ganglion neurons, hence contributing to anti-inflammatory effects.
1.2b channel. Nitro-oleic acid activates TRPA1 and TRPV1 channels in sensory neurons involved in neurogenic inflammation and pain induced by noxious chemicals or thermal stimuli, thereby provoking calcium influx, membrane depolarization, and firing. In addition, high concentrations of nitro-oleic acid suppress firing in dorsal root ganglion neurons, hence contributing to anti-inflammatory effects.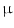 mol concentrations) [124].
mol concentrations) [124].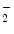 )-derived nitrogen dioxide (NO
)-derived nitrogen dioxide (NO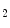 ), these reactions being favored by the prooxidative condition of inflammation. In addition, when pH is low enough (
), these reactions being favored by the prooxidative condition of inflammation. In addition, when pH is low enough (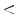 6) to protonate NO
6) to protonate NO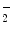 to nitrous acid (HNO
to nitrous acid (HNO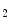 ), this condition also yields the nitrating species NO
), this condition also yields the nitrating species NO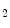 . Therefore, nitrogen dioxide is both a product of oxidative inflammatory reactions involving nitric oxide and nitrite and acidic conditions in the presence of nitric oxide or nitrite. Similarly, nitroalkenes are produced by addition of the radical nitrogen dioxide (NO
. Therefore, nitrogen dioxide is both a product of oxidative inflammatory reactions involving nitric oxide and nitrite and acidic conditions in the presence of nitric oxide or nitrite. Similarly, nitroalkenes are produced by addition of the radical nitrogen dioxide (NO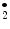 ) to one or more of the olefinic carbons of unsaturated fatty acids [123].
) to one or more of the olefinic carbons of unsaturated fatty acids [123]. -oxidation of odd-chain fatty acids yields propionylCoA, an anaplerotic substrate that, in the liver, can also be packaged into C5 ketone bodies [125]. In extrahepatic cells, C5 ketone bodies are oxidized by CoA transferase SCOT, hence regenerating propionylCoA.
-oxidation of odd-chain fatty acids yields propionylCoA, an anaplerotic substrate that, in the liver, can also be packaged into C5 ketone bodies [125]. In extrahepatic cells, C5 ketone bodies are oxidized by CoA transferase SCOT, hence regenerating propionylCoA. D
D ),7 and fibroblast growth factor FGF23.
),7 and fibroblast growth factor FGF23.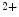 concentration rapidly provokes secretion of PTH that inhibits renal calcium excretion and stimulates renal hydroxylation of 25OH-vitamin-D to calcitriol. The latter triggers intestinal calcium absorption (Table 2.6). In addition, calcitriol and PTH cause bone resorption by osteoclasts. Conversely, hypercalcemia inhibits PTH secretion via parathyroid calcium-sensing receptors.
concentration rapidly provokes secretion of PTH that inhibits renal calcium excretion and stimulates renal hydroxylation of 25OH-vitamin-D to calcitriol. The latter triggers intestinal calcium absorption (Table 2.6). In addition, calcitriol and PTH cause bone resorption by osteoclasts. Conversely, hypercalcemia inhibits PTH secretion via parathyroid calcium-sensing receptors.Organ | Effect | Regulators |
|---|---|---|
Gut | Ca 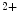 absorption absorption | Calcitriol (⊕) |
Phosphate absorption | Calcitriol (⊕) | |
FGF23 (⊖) | ||
Kidney | Ca 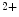 excretion excretion | PTH (⊖) |
Hypercalcemia (⊕) | ||
Phosphate excretion | PTH (⊕) | |
FGF23 (⊕) | ||
Hyperphosphatemia (⊕) | ||
Calcitriol synthesis | PTH (⊕) | |
Hypercalcemia (⊖) | ||
Hyperphosphatemia (⊖) | ||
Bone | Ca 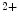 release release | PTH (⊕) |
Calcitriol (⊕) | ||
Phosphate release | PTH (⊕) | |
Calcitriol (⊕) | ||
PTG | PTH secretion | Hypercalcemia (⊖) |
Hyperphosphatemia (⊕) | ||
Calcitriol (⊖) |
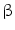 -glucoronidase Klotho coreceptor. Membrane-bound Klotho selectively localizes to the kidney, parathyroid gland, and choroid plexus. The FGF23–Klotho dimer binds to the fibroblast growth factor receptor, a receptor Tyr kinase, thereby causing its autophosphorylation and triggering signaling via 3 major pathways: PI3K–PKB, PLC
-glucoronidase Klotho coreceptor. Membrane-bound Klotho selectively localizes to the kidney, parathyroid gland, and choroid plexus. The FGF23–Klotho dimer binds to the fibroblast growth factor receptor, a receptor Tyr kinase, thereby causing its autophosphorylation and triggering signaling via 3 major pathways: PI3K–PKB, PLC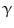 –PKC, and Ras–MAPK. Factor FGF23 regulates phosphate balance via expression of genes involved in PTH, vitamin-D, and phosphate metabolism. In cardiomyocytes, FGF2 uses heparan sulfate proteoglycans as coreceptors, FGFR, and primarily the Ras–MAPK pathway, whereas FGF23 signals primarily via the PLC
–PKC, and Ras–MAPK. Factor FGF23 regulates phosphate balance via expression of genes involved in PTH, vitamin-D, and phosphate metabolism. In cardiomyocytes, FGF2 uses heparan sulfate proteoglycans as coreceptors, FGFR, and primarily the Ras–MAPK pathway, whereas FGF23 signals primarily via the PLC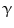 –PP3 pathway.
–PP3 pathway.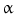 -hydroxylase, hence the renal synthesis of calcitriol [126]. Proximal tubule cells of the nephron produce fibroblast growth factor receptors FGFR1, FGFR3, and FGFR4, FGFR1 being the predominant receptor for the FGF23 hypophosphatemic action [128]. In the proximal tubule, FGF23 reduces production and activity of 2 sodium–phosphate cotransporters NaPi2a and NaPi2c ( SLC34a1 and SLC34a3), hence augmenting phosphaturia. In addition, FGF23 suppresses PTH synthesis in the parathyroid glands.
-hydroxylase, hence the renal synthesis of calcitriol [126]. Proximal tubule cells of the nephron produce fibroblast growth factor receptors FGFR1, FGFR3, and FGFR4, FGFR1 being the predominant receptor for the FGF23 hypophosphatemic action [128]. In the proximal tubule, FGF23 reduces production and activity of 2 sodium–phosphate cotransporters NaPi2a and NaPi2c ( SLC34a1 and SLC34a3), hence augmenting phosphaturia. In addition, FGF23 suppresses PTH synthesis in the parathyroid glands.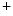 –Pi cotransporters leads to inhibitory phosphorylation of NOS3 synthase.
–Pi cotransporters leads to inhibitory phosphorylation of NOS3 synthase.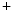 –Pi cotransporters into the cell. Calcium ion stimulates Na
–Pi cotransporters into the cell. Calcium ion stimulates Na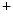 –Pi cotransporter SLC34a1 on vascular smooth myocytes, thereby permitting intracellular accumulation of phosphate ions.
–Pi cotransporter SLC34a1 on vascular smooth myocytes, thereby permitting intracellular accumulation of phosphate ions.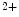 supports liberation of matrix vesicle-like particles of living vSMCs and apoptotic bodies from apoptotic vSMCs that act as nuclei for extracellular calcium–phosphate precipitation [127]. Calcium also reduces the expression of calcification inhibitors by vascular smooth myocytes.
supports liberation of matrix vesicle-like particles of living vSMCs and apoptotic bodies from apoptotic vSMCs that act as nuclei for extracellular calcium–phosphate precipitation [127]. Calcium also reduces the expression of calcification inhibitors by vascular smooth myocytes.2.3.2 Inflammatory Intestine, Gut Microbiome, and Cardiovascular Disease
2.3.2.1 Intestinal Microbiota
2.3.2.2 Oral Drugs and Alimentary Tract
2.3.2.3 Altered Intestinal Barrier and Bacteria
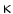 B and transcription of proinflammatory genes. Single nucleotide polymorphism in the Tlr4 gene replacing Asp299 with Gly (D299G) and Thr399 with Ile (T399I) in TLR4 causes lipopolysaccharide hyporesponsiveness. These two genetic variants are linked to various infectious and noninfectious diseases.13 Carriers of 1 or 2 alleles with TLR4 polymorphisms (Asp299Gly and Thr399Ile) are more susceptible to bacterial infections. In particular, patients with the Asp299Gly TLR4 allele have lower levels of certain proinflammatory cytokines (e.g., IL6), acute-phase reactants, and adhesion molecules. On the other hand, the Asp299Gly TLR4 polymorphism that attenuates TLR4 signaling and inflammatory response to Gram− bacteria decreases risk of atherosclerosis [134].
B and transcription of proinflammatory genes. Single nucleotide polymorphism in the Tlr4 gene replacing Asp299 with Gly (D299G) and Thr399 with Ile (T399I) in TLR4 causes lipopolysaccharide hyporesponsiveness. These two genetic variants are linked to various infectious and noninfectious diseases.13 Carriers of 1 or 2 alleles with TLR4 polymorphisms (Asp299Gly and Thr399Ile) are more susceptible to bacterial infections. In particular, patients with the Asp299Gly TLR4 allele have lower levels of certain proinflammatory cytokines (e.g., IL6), acute-phase reactants, and adhesion molecules. On the other hand, the Asp299Gly TLR4 polymorphism that attenuates TLR4 signaling and inflammatory response to Gram− bacteria decreases risk of atherosclerosis [134].2.3.2.4 Inflammatory Bowel and Risk for Cardiovascular Diseases
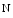 oxide, and betaine, predicts risk for cardiovascular disease [132].
oxide, and betaine, predicts risk for cardiovascular disease [132].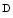 xylose lowers. In addition, the bacterial density in the sigmoidal mucosal biofilm and the extent of their adherence heighten with respect to healthy subjects.
xylose lowers. In addition, the bacterial density in the sigmoidal mucosal biofilm and the extent of their adherence heighten with respect to healthy subjects.2.3.3 Psychological Context
2.3.4 Smoking
 consumption. An increased resting cardiac frequency reduces cardiac performance and ischemic threshold.
consumption. An increased resting cardiac frequency reduces cardiac performance and ischemic threshold.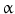 1-antitrypsin), which is carried by high-density lipoproteins at low levels in dyslipidemia, among other factors.
1-antitrypsin), which is carried by high-density lipoproteins at low levels in dyslipidemia, among other factors.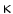 B activation. As with other cardiovascular risk factors (diabetes, dyslipidemia, hypertension, and renal disease), smoking is associated with a decreased number and function of bone marrow-derived endothelial progenitor cells, which participate in endothelial regeneration and angiogenesis. Smoking (1) increases levels of proinflammatory compounds (e.g., tumor-necrosis factor-
B activation. As with other cardiovascular risk factors (diabetes, dyslipidemia, hypertension, and renal disease), smoking is associated with a decreased number and function of bone marrow-derived endothelial progenitor cells, which participate in endothelial regeneration and angiogenesis. Smoking (1) increases levels of proinflammatory compounds (e.g., tumor-necrosis factor-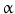 and endothelial intercellular adhesion molecule-1), and reactive oxygen species, (2) decreases concentrations of anti-inflammatory, antioxidant HDL–cholesterol (HDL
and endothelial intercellular adhesion molecule-1), and reactive oxygen species, (2) decreases concentrations of anti-inflammatory, antioxidant HDL–cholesterol (HDL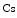 ), which promotes reverse cholesterol transport, and adiponectin, and (3) activates leukocytes and platelets.
), which promotes reverse cholesterol transport, and adiponectin, and (3) activates leukocytes and platelets.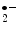 ) as well as peroxynitrite (ONOO
) as well as peroxynitrite (ONOO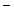 ) and causes oxidation (inactivation) of nitric oxide NOS3 synthase [138]. Smokers have a drop of endothelium-mediated vasodilation.
) and causes oxidation (inactivation) of nitric oxide NOS3 synthase [138]. Smokers have a drop of endothelium-mediated vasodilation.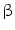 , and tissue factor [140].
, and tissue factor [140].2.3.5 Resting Tachycardia
Afferents and central processors | |
1 | Arterial baroreceptors |
2 | Excitatory (glutamatergic) afferents neurons |
3 | Excitatory (glutamatergic) neurons of NTS |
4 | Inhibitory (gabaergic) neurons of CVLM |
5 | Excitatory (glutamatergic) neurons of RVLM |
Efferents | |
6 | Sympathetic preganglionic (cholinergic) neurons of ILNSC |
7 | Sympathetic postganglionic (noradrenergic) neurons |
8 | Adrenergic receptors of vascular endothelial and smooth muscle cells |
Adrenergic receptors of cardiac cells | |
Adrenergic receptors of renal cells | |
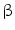 2-adrenergic receptor raises endothelial cytosolic concentration of free calcium ion and NOS3-dependent NO production and release.
2-adrenergic receptor raises endothelial cytosolic concentration of free calcium ion and NOS3-dependent NO production and release. 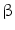 1-adrenoceptor does not reside on endothelial cells [143].
1-adrenoceptor does not reside on endothelial cells [143].2.3.6 Hypertension
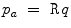 ) is controlled by peripheral vascular resistance (
) is controlled by peripheral vascular resistance (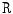 ) and blood flow rate (q).15 Vascular resistance depends on the vasomotor tone under local and remote control. Blood flow rate depends on cardiac performance and blood volume, itself related to salt content.
) and blood flow rate (q).15 Vascular resistance depends on the vasomotor tone under local and remote control. Blood flow rate depends on cardiac performance and blood volume, itself related to salt content.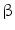 -adrenergic and angiotensin AT
-adrenergic and angiotensin AT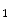 receptors abolish the action of adrenaline and angiotensin-2.
receptors abolish the action of adrenaline and angiotensin-2. 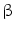 -blockers inhibit
-blockers inhibit 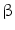 -adrenergic receptors, thereby decreasing cardiac frequency and contractility.
-adrenergic receptors, thereby decreasing cardiac frequency and contractility. 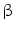 -adrenoceptors initiate signaling cascades upon phosphorylation by AMPK, CamK2, PKA, PKB, and TOR kinases. These enzymes target in particular ion channels such as K
-adrenoceptors initiate signaling cascades upon phosphorylation by AMPK, CamK2, PKA, PKB, and TOR kinases. These enzymes target in particular ion channels such as K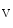 7.1 and transporters that mediate ion fluxes at high cardiac frequency [144]. G-protein-coupled receptors that regulate myocardial contractility are also substrates. Inhibitors of angiotensin-converting enzyme prevent formation of angiotensin-2. Diuretics hinder increase in plasma volume. Calcium antagonists treat vasoconstriction.
7.1 and transporters that mediate ion fluxes at high cardiac frequency [144]. G-protein-coupled receptors that regulate myocardial contractility are also substrates. Inhibitors of angiotensin-converting enzyme prevent formation of angiotensin-2. Diuretics hinder increase in plasma volume. Calcium antagonists treat vasoconstriction.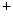 –K
–K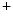 ATPase triggers Ca
ATPase triggers Ca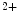 -dependent and -independent pathways.
-dependent and -independent pathways.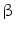 , and IP
, and IP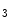 , thereby inducing Ca
, thereby inducing Ca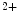 release from intracellular stores and myosin light chain phosphorylation by activation of myosin light chain kinase.
release from intracellular stores and myosin light chain phosphorylation by activation of myosin light chain kinase.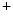 –K
–K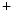 ATPase and launch Ca
ATPase and launch Ca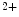 influx, as well as inhibit myosin light chain dephosphorylation via Rho GTPase to prevent smooth muscle relaxation.
influx, as well as inhibit myosin light chain dephosphorylation via Rho GTPase to prevent smooth muscle relaxation.2.3.7 Diabetes
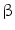 cells of islets of Langerhans in the pancreas. Type-2 diabetes (a.k.a. insulin-independent, obesity-related, and adult-onset diabetes) results from insulin resistance, as cells do not respond to the insulin. Sometimes, it combines resistance to insulin action, inadequate insulin secretion, and inappropriate glucagon secretion. Gestational diabetes occurs when pregnant women develop hyperglycemia. Classical symptoms include polyuria, polydipsia, and polyphagia. Long-term complications include microangiopathy, diabetic neuropathy, chronic renal failure, diabetic retinopathy, and cardiovascular disease such as aggravated atherosclerosis.
cells of islets of Langerhans in the pancreas. Type-2 diabetes (a.k.a. insulin-independent, obesity-related, and adult-onset diabetes) results from insulin resistance, as cells do not respond to the insulin. Sometimes, it combines resistance to insulin action, inadequate insulin secretion, and inappropriate glucagon secretion. Gestational diabetes occurs when pregnant women develop hyperglycemia. Classical symptoms include polyuria, polydipsia, and polyphagia. Long-term complications include microangiopathy, diabetic neuropathy, chronic renal failure, diabetic retinopathy, and cardiovascular disease such as aggravated atherosclerosis. arginine–nitric oxide pathway is activated [146]. Second messengers involved in adenosine signaling include PKC, ERK1, and ERK2 that activate
arginine–nitric oxide pathway is activated [146]. Second messengers involved in adenosine signaling include PKC, ERK1, and ERK2 that activate 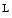 arginine ingress through SLC7a1,18 but preclude adenosine import through the SLC29a1 carrier.19 Subsequent extracellular accumulation of adenosine activates the A
arginine ingress through SLC7a1,18 but preclude adenosine import through the SLC29a1 carrier.19 Subsequent extracellular accumulation of adenosine activates the A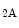 receptor, increases transcription of the Nos3 and SLC7A1 genes, hence NO synthesis. Cultured human umbilical vein endotheliocytes from gestational diabetic pregnancies or subjected to hyperglycemia produce higher NO levels. However, the NO-dependent downregulation of the SLC29A1 gene transcription lowers uptake of vasodilatory adenosine [146]. Nitric oxide supports formation of the complex made of DNA-damage-inducible transcript DDIT3, or CCAAT/enhancer-binding protein (C/EBP) homologous protein-10 (CHOP or CHP10),20 and C/EBP
receptor, increases transcription of the Nos3 and SLC7A1 genes, hence NO synthesis. Cultured human umbilical vein endotheliocytes from gestational diabetic pregnancies or subjected to hyperglycemia produce higher NO levels. However, the NO-dependent downregulation of the SLC29A1 gene transcription lowers uptake of vasodilatory adenosine [146]. Nitric oxide supports formation of the complex made of DNA-damage-inducible transcript DDIT3, or CCAAT/enhancer-binding protein (C/EBP) homologous protein-10 (CHOP or CHP10),20 and C/EBP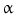 factor [147].21 In the nucleus, like the transcription factor Specific protein SP1 in hyperglycemia, the DDIT3–C/EBP
factor [147].21 In the nucleus, like the transcription factor Specific protein SP1 in hyperglycemia, the DDIT3–C/EBP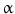 heterodimer represses Slc29a1 gene transcription (as well as glucose transporter GluT4) [147].
heterodimer represses Slc29a1 gene transcription (as well as glucose transporter GluT4) [147].2.3.8 Metabolic Syndrome
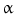 , in addition to IL1
, in addition to IL1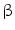 , IL4, IL8, and IL18, or anti-inflammatory such as interleukin-10; (4) serpins; such as serpin-E1, or plasminogen activator inhibitor PAI1, and serpin-F1, or pigment epithelium-derived factor (PEDF); (5) fatty acid-binding proteins (FABPs) such as adipocyte FABP (aFABP, or FABP4); (6) lipocalin-2; and (7) adhesion glycoproteic molecules such as thrombospondin-1.
, IL4, IL8, and IL18, or anti-inflammatory such as interleukin-10; (4) serpins; such as serpin-E1, or plasminogen activator inhibitor PAI1, and serpin-F1, or pigment epithelium-derived factor (PEDF); (5) fatty acid-binding proteins (FABPs) such as adipocyte FABP (aFABP, or FABP4); (6) lipocalin-2; and (7) adhesion glycoproteic molecules such as thrombospondin-1.Process | Pathway and effect |
|---|---|
Angiogenesis | VEGF |
Apoptosis | AMPK, ceramidase–S1P Inhibition of caspase-8 |
Glucose uptake | PKB GluT4 transfer to the plasma membrane |
Inflammation | SphK1–COx2 Inhibition of TNFSF1 |
Lipid uptake | AMPK–ScaRb3 |
Oxidative and nitrative stresses | Inhibition of NOS2 Inhibition of NOx2 |
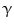 or endotoxin promote interleukin-12-mediated helper T
or endotoxin promote interleukin-12-mediated helper T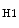 lymphocyte-based immunity. Activated macrophages by IL4 or IL13 (M2a), immune complexes (M2b), and anti-inflammatory cytokines IL10 or TGF
lymphocyte-based immunity. Activated macrophages by IL4 or IL13 (M2a), immune complexes (M2b), and anti-inflammatory cytokines IL10 or TGF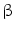 (M2c) support T
(M2c) support T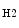 -based immunity implicated in wound healing, tissue repair, and resolution of inflammation. Obesity favors macrophage polarization to the proinflammatory M1 phenotype [150]. Kinase JNK is required for the differentiation of proinflammatory macrophages. In myeloid cells, JNK1 and JNK2 are involved in obesity-induced recruitment of macrophages and inflammation in adipose tissue, without affecting other myeloid cell populations (eosinophils and neutrophils) [150].
-based immunity implicated in wound healing, tissue repair, and resolution of inflammation. Obesity favors macrophage polarization to the proinflammatory M1 phenotype [150]. Kinase JNK is required for the differentiation of proinflammatory macrophages. In myeloid cells, JNK1 and JNK2 are involved in obesity-induced recruitment of macrophages and inflammation in adipose tissue, without affecting other myeloid cell populations (eosinophils and neutrophils) [150].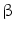 cells, increases the synthesis and storage of triglycerides in hepatocytes, and decreases ATP generation both from carbohydrate and fatty acid oxidation in skeletal muscle [152].
cells, increases the synthesis and storage of triglycerides in hepatocytes, and decreases ATP generation both from carbohydrate and fatty acid oxidation in skeletal muscle [152].2.3.9 Altered Sleep
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


