Figure 27–1.
Compendium of LQTS-susceptibility genes
Rare subtypes stem from perturbations in key cardiac channel interacting proteins [Kv7.1, Kv11.1 or Nav1.5 beta subunits (LQT5, LQT6, LQT10, LQT11, LQT12) or structural membrane scaffolding proteins such as ankyrin B (LQT4) or caveolin 3 (LQT9)]. The latest edition, LQT13, identified in 2010 in a single large Chinese pedigree with concomitant prolonged QTc and persistent atrial fibrillation, stems from mutations in the KCNJ5-encoded Kir3.4 potassium channel [10].
Complex, multi-system syndromes that include abnormal repolarization have been included as variants of LQTS. These syndromes include Andersen Tawil syndrome (ATS1, also annotated as LQT7) due in part to mutations in the KCNJ2-encoded Kir2.1/IK1 potassium channel and Timothy syndrome (TS1, also annotated as LQT8) due to mutations in the L-type calcium channel (Cav1.2) alpha subunit. This chapter will focus mostly on the clinically pure forms of LQTS, particularly the three most common subtypes, LQT1-3.
Clinical Presentations
The clinical manifestation of LQTS ranges from a lifelong asymptomatic course in some cases to premature sudden cardiac death (SCD) during infancy in others. As a simple rule of thumb, approximately 50 % of patients with genetic evidence of LQTS will never have a symptom attributable to LQTS and a significant proportion (ranging from 10 to 40 % depending on genotype) of those with genetic LQTS do not show overt QT prolongation [5, 11, 12]. Patients failing to display QT prolongation at rest are said to have Concealed LQTS or Normal QT Interval LQTS. While risk of a LQTS-precipitated event generally increases with increasing QTc, it is nevertheless possible to experience SCD as a sentinel event despite having Concealed LQTS (i.e. normal resting QTc). This is fortunately quite uncommon. The most common presenting symptoms are recurrent syncope, seizures, and sudden cardiac death (Fig. 27.2). These events are due to the hallmark feature of LQTS, namely polymorphic ventricular tachyarrhythmias called Torsade des Pointes (TdP) which are most often self terminating. Rarely, TdP continue to degenerate, yielding ventricular fibrillation and sudden death. Overall, less than 5 % of patients with LQTS will present with a sentinel event of sudden death or aborted cardiac arrest. Conversely, nearly half of LQTS sudden death victims experienced a prior warning episode of syncope.
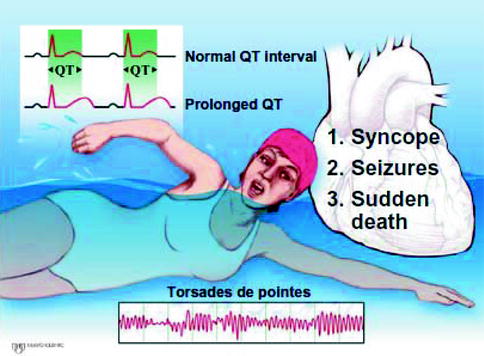
Figure 27–2.
Summary of the key clinical features of LQTS
Syncope is the most frequent symptom occurring commonly between age 5 and 15 years. Among symptomatic probands, 50 % experience their first cardiac event by 12 years of age (particularly boys), and by 40 years of age the proportion increases to almost 90 % [13]. In general, approximately 50 % of patients present with activity – or emotion-related symptoms – primarily syncope, seizures, or palpitations. The majority of cardiac events are related to physical activity or emotional stress. To be certain, a “fight-flight-fright”-triggered faint must be considered potentially ominous until proven otherwise and a meticulous LQTS evaluation must be conducted. In fact, data from the LQTS Registry indicates that recent syncope is the most significant harbinger of future sudden death, greater than genotype and degree of QT prolongation [14]. Thus, it is critically important to properly distinguish an ordinary vasovagal faint from a possibly torsadogenic one.
In general and without consideration of the underlying LQTS-causing genotype, the risk of the first cardiac event in males is typically higher in childhood and decreases after puberty, perhaps due in part to regression of QTc duration [15, 16]. On the other hand, during adolescence and adulthood, females appear more vulnerable to LQTS-related cardiac events. In addition, females are at significant risk for cardiac events during the postpartum period. In fact, Rashba and colleagues reported nearly 10 % of female probands experienced their first cardiac events during the postpartum period [17].
Genotype-Phenotype Specific Correlations
Some of the phenotypic heterogeneity in LQTS is now understood because of the underlying genetic heterogeneity particular with respect to gene-specific triggers for cardiac events [18]. Figure 27.3 summarizes some of the genotype-specific features observed in LQT1, LQT2 and LQT3.
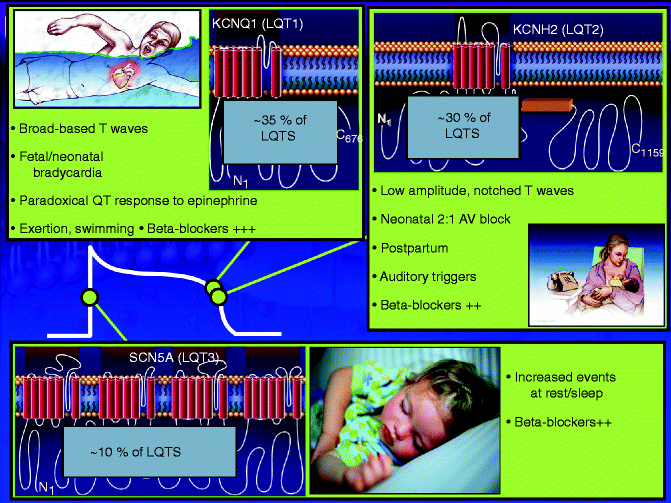
Figure 27–3.
Summary of key genotype-phenotype correlations (Modified from Tester and Ackerman [19] with permission from Elsevier)
LQT1 Phenotype
Patients with the most common genetic subtype (LQT1) predominantly have exertional-triggered symptoms [18]. Swimming is a relatively gene-specific arrhythmogenic trigger associated almost exclusively with LQT1 [20, 21]. With few exceptions to date, all LQTS patients, with either a personal history or an extended family history of a near drowning, have a defective KCNQ1 gene facilitating strategic genotyping [21]. The common triggers other than swimming include running, startle, anger and fright. LQT1 mutations cause defective IKs channel which is responsive to adrenergic stimulation, so the usual shortening of QT in response to increased heart rate is impaired and QTc progressively lengthens during exercise and early recovery. The common ECG finding in LQT1 is prolonged T wave duration or a broad based T wave pattern [22, 23].
LQT2 Phenotype
Auditory stimuli, such as a telephone ringing or an alarm clock sounding, is a common trigger in LQTS and often indicates the presence of a KCNH2 (LQT2) defect [24]. Additionally, there is a relatively gene-specific molecular basis underlying cardiac events during the postpartum period in LQTS [25]. Postpartum cardiac events were found more common in LQT2 (16 %) than in LQT1 (<1 %) [25, 26]. Fifteen percent of LQT2-associated cardiac events occur during rest or sleep. The finding of bifid T waves in the inferior and lateral leads is characteristic of LQT2 [27].
LQT3 Phenotype
Sleep/rest-triggered events seem most common in patients harboring a defective sodium channel Nav1.5 encoded by SCN5A [18, 28]. In LQT3, SCN5A mutations impair inactivation of INa, causing repetitive re-openings throughout the action potential and persistent inward current, resulting in prolongation of the action potential duration and QT interval [29].
The underlying genotype has a profound influence on the clinical course [30]. LQT1 and LQT2 families comprise approximately 70 % of LQTS and have a much higher risk of cardiac events than patients with LQT3. However, the “lethality” of a given cardiac event appears to be greatest in LQT3. Fortunately, SCN5A-based LQTS (LQT3) is approximately ten-fold less common than the potassium channel LQTS subtypes.
Diagnostic Evaluation
Schwartz et al. proposed the first diagnostic criteria for LQTS in 1985, that included the following major criteria: QTc > 440 ms, stress induced syncope, family members with LQTS, and minor criteria: congenital deafness, episodes of T-wave alternans, low heart rate (in children) and abnormal ventricular repolarization [31]. However, the more recent understanding of the “overlap zone” between LQTS and health has rendered this cut-off value of 440 ms a major limitation of the original “Schwartz criteria.”
A modified “Schwartz score” containing new criteria and a point system based upon a range of QTc values and the clinical/family history was formulated in 1993 and in 2011, the diagnostic utility of the QTc at the 3rd/4th minute of recovery from exercise stress testing was added (Table 27.1) [32, 33]. The “Schwartz score”, recently modified for what concerns the points necessary for a “high clinical probability of LQTS” ranges from 0 to 9 and contains three diagnostic probabilities: ≤1 point, low probability of LQTS; 2 or 3 points, intermediate probability of LQTS; and ≥3.5 points, high probability of LQTS [34]. The Schwartz criteria provide a useful guide for contemplating a clinical diagnosis of LQTS with the positive predictive value of a modified “Schwartz score” ≥3.5 approaching 100 %.
Table 27.1
Schwartz/Moss score for LQTS diagnostic criteria
Variable | Points |
|---|---|
ECG findings | |
QTc msa | |
≥480 | 3 |
460–479 | 2 |
450–459 (in males) | 1 |
QTca fourth minute of recovery from exercise stress test ≥480 ms | 1 |
Torsades de pointesb | 2 |
T-wave alternans (macroscopic) | 1 |
Notched T wave in three leads | 1 |
Low heart rate for ageb | 0.5 |
Clinical history | |
Syncopec | |
With stress | 2 |
Without stress | 1 |
Congenital deafness | 0.5 |
Family history d | |
Family members with definite LQTSe | 1 |
Unexplained sudden cardiac death < age 30 years among immediate family members | 0.5 |
The Schwartz criteria should be used to reasonably select the candidates for genetic testing; in turn, the results should be used to identify by cascade screening the “silent” mutation positive family members. By definition, these criteria CAN NOT identify the patients or family members with Concealed LQTS. This critical point stems from the observation that a significant proportion of LQTS is concealed and that the penetrance, defined as the ratio between patients with a manifest clinical phenotype (QT prolongation, symptoms, etc.) and the total number of family members who are positive for the mutation identified in the LQTS proband may be as low as 25 %. Although the clinical risk of the patient with Concealed LQTS is much lower than the patient with manifest LQTS, the risk of an untoward cardiac event is not zero and hence, it is essential to correctly identify all family members. Thus, in the evaluation of first degree relatives of a definitely affected LQTS proband, it is no longer acceptable to exclude LQTS among individuals based upon an equivocal or even normal QTc or a low Schwartz score. Instead, genotyping, for the estimated 75 % of families who possess an LQTS-causing mutation, is the only definitive diagnostic test for the rest of the family.
Symptomatic patients, who have an ECG with a so-called borderline QTc (440–460 ms) or normal QTc, will have a borderline or questionable (score of 2 or 3) LQTS diagnosis based on clinical criteria. In this situation, serial ECGs should be performed since the QTc value in LQTS patients may vary from time to time. Furthermore, a careful inquiry about family history of sudden death as well as screening ECGs from other family members may be informative. In addition, stress testing using exercise protocols or the epinephrine QT stress test may be helpful in exposing LQTS, particularly LQT1 [33, 35], and a simple brisk “stand-up” test may be useful in unmasking LQT1 and LQT2 [36].
The 12-Lead Electrocardiogram
The 12-lead ECG remains the cornerstone in the LQTS clinical diagnosis and the principal screening tool. The hallmark ECG feature of LQTS includes prolongation of the rate-corrected QT interval (QTc) as measured by Bazett’s formula (QTc = QT interval/square root of RR interval) [37]. The QT interval is defined as the time interval between the onset of QRS and the end of the T wave. This value is corrected for the heart rate by dividing it by the square root of the preceding RR interval (Bazett’s QTc formula). Note, with a heart rate of 60 beats per minute (RR interval = 1 s), the QTc equals the QT interval. More rapid heart rates cause the calculated QTc to increase relative to the measured QT interval.
Lead II is generally the accepted lead for QTc calculation because the inscription of the T wave is usually discrete [38]. Alternatively, the lateral precordial leads V5 and V6 are sometimes quite informative [38, 39]. When sinus arrhythmia is present, an average QTc from the entire lead II strip (at least three consecutive determinations) must be determined [28, 40]. Simply taking the longest observed QTc will result in too many false positive classifications.
It is vital that the ECG be visualized directly and the QTc manually calculated by a physician with expertise in LQTS. The computer calculated QTc is NOT acceptable in the evaluation of LQTS. Independent manual calculation is mandatory. Unfortunately, Viskin and colleagues have exposed that humans are not much better than the computer, in fact, humans are worse, when it comes to accurately calculating the QT interval. Compared to “QT aficionados” who made significant QTc miscalculations <5 % of the time, heart rhythm specialists and general cardiologists often failed to correctly calculate the QTc (>60 and >75 % of the time respectively) [41].
Even when the QTc has been calculated accurately, the abundance of Concealed LQTS and misconceptions about the normal distribution of the QTc pose a serious diagnostic challenge (Fig. 27.4). Before the molecular revelations in LQTS, a QTc of ≥440 ms (males) and ≥460 ms (females) was considered prolonged [42]. Subsequently, Vincent et al. [28] examined the QTc distribution in three families with LQT1 and found that no genotype positive individuals had a QTc ≤ 410 ms and no unaffected (genotype negative) persons had a QTc ≥ 470 ms in men or ≥480 ms in women, and that a significant number of patients were classified erroneously using the 440 ms cut-off. From a screening standpoint, if a QTc of 460 ms is used as a cutoff point, the positive predictive value is 92 % and the negative predictive value is 94 % [5]. Again for screening purposes, we advocate designating QTc values ≥470 ms during infancy or ≥500 ms in adolescents/adults as “prolonged” to maximize both the positive and negative predictive values as a screening test.
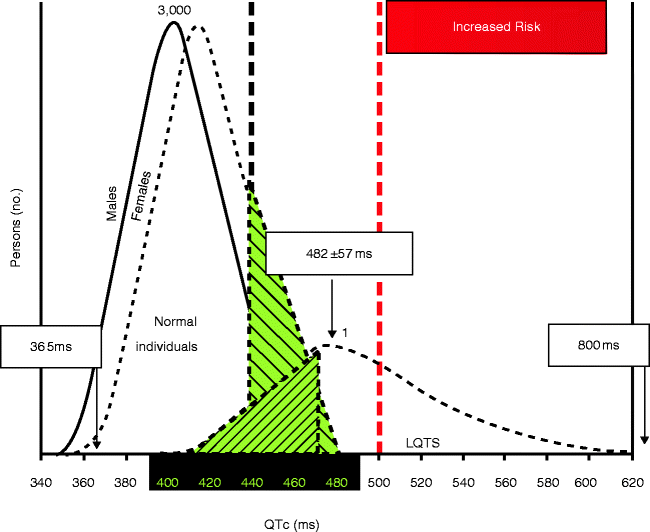
Figure 27–4.
QTc distribution in health and LQTS
Figure 27.4 depicts the distribution of QTc values among healthy postpubertal adult men and women as well as the QTc distribution seen for all genotype positive patients evaluated in Mayo Clinic’s LQTS clinic. Despite our “indoor track record” of a genotyped LQT1 family member with a QTc of 365 ms, the interpretation of a QTc < 400 ms is statistically easy – Normal, no LQTS – with virtually 100 % negative predictive value. On the other end, a QTc ≥ 500 ms almost always indicates repolarization pathology due to either acquired or congenital LQTS. More importantly, the overlap zone (400–480 ms) is where great consternation occurs. Here, the physician must understand the normal QTc distribution in healthy subjects versus the 1 in 2,500 LQTS-affected persons. For example, an asymptomatic patient with a negative family history and a screening QTc of 450 ms has >500:1 odds favoring normalcy rather than happening to catch someone with Concealed LQTS.
Complicating matters further is the observation that LQTS individuals with a “normal” ECG (i.e. “low-penetrant” LQTS or “concealed” LQTS) can indeed, albeit uncommonly, have cardiac events including sudden death. Approximately 10 % of genotype positive index cases presented for LQTS genetic testing with a QTc ≤ 440 ms [43]. These observations have transformed this extensive “overlap zone” into the “nightmare zone.” Thus, although the risk for sudden death in a patient with a QTc < 440 ms remains exceedingly rare, the presence of a normal QTc in a genotype positive LQTS patient must NOT be interpreted as a risk free marker. As will be discussed later in the treatment section, such genotype positive/phenotype negative patients must be advised of simple, potentially life saving preventative measures including avoidance of medications that prolong the QT interval. Understanding this QTc overlap zone is foundational to the LQTS evaluation and constitutes one of the major misunderstandings in the clinical evaluation of LQTS.
T-U Wave Abnormalities and T Wave Alternans
Besides manually calculating the QTc and understanding the QTc overlap zone, sleuth-like inspection of the morphology of the T waves should also be conducted in a LQTS evaluation (Fig. 27.5). Unusual T waves – wide-based slowly generated, notched, bifids, biphasic, low amplitude humps and bumps on the down-slope limb, indistinct termination due to U waves, sinusoidal oscillation, or simply a delayed inscription of normally appearing T waves may lead to the diagnosis of LQTS despite a normal or borderline QTc [22]. These T wave abnormalities may be evident particularly in the lateral precordial leads. However, caution must be exercised with concluding LQT-suspicious T wave peculiarities based upon changes in the precordial leads of V2 and V3. In addition, the T wave morphology may be somewhat gene-specific providing another piece of evidence permitting strategic genotyping [22, 44]. Patients with LQT3 tend to have a late-appearing T wave clearly distinct from the low-amplitude, moderately delayed T wave observed in LQT2. Both of these T-wave profiles are different from the broad-based, prolonged T-wave pattern seen in LQT1 [22, 23]. However, the classic LQT3-like T wave pattern can be seen often in patients with LQT1.
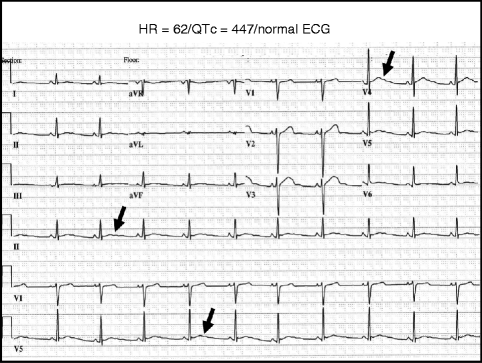
Figure 27–5.
Example of T-wave sleuthing – 12-lead ECG in proband with LQT2, borderline QT prolongation, and abnormal T wave morphology
In 1994, Malfatto and colleagues provided the first evidence that morphological analysis of T wave abnormalities, namely notched or biphasic T waves, may contribute to the diagnosis and risk stratification of LQTS. Herein, notched or biphasic T waves were present in 62 % of LQTS patients compared to only 15 % of control subjects (p < 0.001). Moreover, 81 % of symptomatic patients compared to 19 % of asymptomatic patients had notched or biphasic T waves (p < 0.001). This same distribution was also observed within LQTS families, in which symptomatic members had more pronounced T wave abnormalities then their asymptomatic family members. In symptomatic patients, the occurrence of T wave abnormalities was independent of the length of repolarization (QTc) [45].
Next, Lupoglazoff and colleagues classified a notched T wave as a grade 1 (G1) notch when it occurred at or below the apex whatever the amplitude, and as grade 2 (G2) when the protuberance occurred above the apex [46]. G2 notches appear more specific and often indicate LQT2. However, baseline ECG showing G2 notches is not found commonly. Recently, Khositseth et al. [47] reported G2 notching elicited during low-dose epinephrine may unmask some patients with concealed LQT2. The mandate for careful inspection of T wave morphology is two-fold: (1) unmask a patient with concealed LQTS – i.e. a normal resting QTc with a peculiar T wave and (2) provide a possible starting point for the mutational analysis based upon the suggested ECG pattern.
Besides QTc and T wave morphology, macrovoltage and microvoltage T wave alternans (TWA) and QT dispersion (QTd) may be informative. TWA is characterized by beat-to-beat alternation of the morphology, amplitude, and/or polarity of the T wave and is a marker of major electrical instability and regional heterogeneity of repolarization and is likely to be associated with an increased risk of cardiac events [48, 49]. QTd is defined as the difference between the maximal and minimal QT intervals in the 12 standard leads and may reflect spatial repolarization [50]. It has been described as an arrhythmic marker for LQTS. There is evidence that QT dispersion is increased in LQTS patients compared to normal controls [51, 52]. Moenning and colleagues have confirmed the finding that a significant and independent difference in QT dispersion between mutation carriers and unaffected family members exists [53]. However, a cut-off value of QT dispersion to distinguish LQTS from health is not available.
Holter Monitoring
In patients with a non-diagnostic QTc, Holter monitoring may aid in the evaluation of LQTS. Again, caution must be exercised with interpreting Holters in a patient with an equivocal history and a borderline QTc. Presently, the normal distribution of 24-h maximal QTc values is poorly understood and concerns have been raised regarding the filter settings in Holter recordings and hence, the precision/accuracy of Holter QT intervals. Critically, a Holter–recorded maximum QTc exceeding 500 ms does not equal LQTS. Instead, the value of repeated Holter recordings lies in capturing the appearance of T-wave patterns (such as T wave notching) that might suggest LQTS in patients with borderline QT prolongation and an uncertain clinical diagnosis [46].
Exercise Testing
Exercise testing may enhance the diagnostic accuracy of the LQTS evaluation as inadequate shortening of QTc with increasing heart rate has been observed [54]. Swan et al. [55] studied 19 LQTS patients and 19 healthy controls undergoing exercise testing. During the recovery phase of exercise, the QT interval lengthened abnormally and the inhomogeneity of repolarization increased in LQTS. LQT2 patients had a lesser degree of QT interval shortening than LQT3 patients in response to increasing heart rate [56]. Moreover, LQT1 patients displayed a diminished chronotropic response and exaggerated prolongation of the QT interval after exercise. In contrast, the QT interval shortens more in LQT2 than in LQT1 patients when heart rate increases and the sinus nodal rate response is normal [57]. The majority of these studies have been conducted in LQTS patients having a diagnostic QTc at rest. Provocative testing is most needed, however, in exposing the patient with concealed LQTS. Here, exercise testing is capable of unmasking patients with concealed LQT1 by detecting inadequate QT adaptation during heart rate increase and especially during the recovery phase [58].
Besides the Ackerman study that demonstrated the ability of the recovery phase QTc, at 2–4 min recovery, to unmask LQT1 whether or not there was QT prolongation at rest and even in the setting of beta blocker therapy [58], Sy and colleagues also proposed a simple 3-step algorithm that incorporates resting ECG, 4-min recovery QTc, and 1-min recovery QTc for the identification of LQTS in asymptomatic relatives and the discrimination between LQT1 and LQT2 [35]. In a derivation cohort of 69 asymptomatic first-degree relatives of LQTS probands (28 with LQT1, 20 with LQT2, and 21 mutation negative), the combination of resting and 4-min recovery QTc (QTc ≥445 ms) in a screening algorithm yielded a sensitivity of 0.94 and specificity of 0.90 for detecting individuals with LQTS mutations. When applied to a validation cohort of 152 LQTS relatives (58 LQT1, 61 LQT2, and 33 mutation negative), the sensitivity was 0.92 and specificity was 0.82. In terms of predicting genotype, the abnormal prolongation of 1-min recovery QTc ≥ 460 ms correctly differentiated LQT1 subtype in 86 of 115 patients (overall accuracy) in their validation cohort, with a sensitivity of 0.73 and specificity of 0.76. This 3-step algorithm was also applied to an independent cohort of 45 possible LQTS probands that were confirmed subsequently to have disease-causing mutations in KCNQ1 or KCNH2. In this third cohort, among patients with a normal or borderline resting QTc, a 4-min recovery QTc ≥ 445 ms correctly identified 25 of 28 patients as having LQTS. The combined diagnostic algorithm had an overall sensitivity of 0.93 for identifying mutation-positive probands [35].
These recent studies provide the general cardiologist with an easily employable tool to be used whenever a LQTS case is suspected [35, 58]. While there is some value in using this algorithm for the identification of asymptomatic relatives of genotyped LQTS probands, the real promise is the potential for confirming the clinical suspicion of LQTS in an ungenotyped borderline diagnosed LQTS patient, where initial genetic testing remains a lengthy process yet there is an urgency to determine if the patient is truly affected so that appropriate prophylactic therapy may be initiated [33, 58]. In fact, given the clinical value delineated in these two studies [35, 59], the evaluation of the recovery phase of exercise has been added to the latest Schwartz score LQTS diagnostic criteria [33].
It is important to note that exercise-induced ventricular ectopy is uncommon in LQTS [59]. In fact, induction of premature ventricular contractions in bigeminy or as couplets and of course, bi-directional ventricular tachycardia, should prompt suspicion for a mimicker of LQTS, namely catecholaminergic polymorphic ventricular tachycardia.
Macrovoltage TWA at rest or during exercise/epinephrine QT stress testing is abnormal and prognosticates increased risk. However, exercise-induced macrovoltage TWA is extremely rare and we have seen epinephrine-induced macrovoltage TWA in only 1 of the past 600 epinephrine QT stress tests [60]. On the other hand, after conducting nearly 100 μV T wave studies using either the epinephrine QT stress test or treadmill stress testing, we cannot assign any clinical utility to this test. In fact, we have discontinued microvoltage T wave testing in both of our Long QT Syndrome Clinics.
Epinephrine QT Stress Test
The epinephrine QT stress test can also unmask concealed LQTS, particularly LQT1. Whether using the Shimizu protocol or the Ackerman/Mayo protocol, the chief finding that points to LQT1 involves a paradoxical lengthening of the QTc (Shimizu protocol) or the absolute QT interval (Ackerman/Mayo protocol) during infusion of epinephrine [60, 61]. When present, a tentative diagnosis of LQT1 is rendered (75 % positive predictive value) and beta blocker therapy is initiated while waiting for confirmation by genetic testing [62]. Given a 96 % negative predictive value, this test is also performed in patients hosting a novel LQT1-associated mutation to provide an in vivo physiological challenge of the patient’s IKs pathway. Such testing provides independent conformation for the pathogenicity of the newly discovered mutation. Importantly, beta blockers significantly confound the interpretation of the epinephrine QT stress test and therefore, unlike the treadmill stress test, it should not be performed in a patient already taking beta blockers. The epinephrine QT stress test is as safe as exercise testing in terms of inducing a dangerous arrhythmia. In over 10 years and over 600 epinephrine QT stress tests conducted at Mayo Clinic, epinephrine-induced torsades has occurred only once. The reader is directed to the Provocative (Drug) Testing in Inherited Arrhythmias chapter by Shimizu and Ackerman which details catecholamine stress testing in the evaluation heritable arrhythmia syndromes with particular focus on the epinephrine QT stress test.
Echocardiographic Analysis by Tissue Doppler Imaging
Echocardiographic analysis by tissue Doppler imaging (TDI) may be useful tool in risk stratification in LQTS. In the early 1990s, Schwartz and colleagues illustrated for the first time, a relationship between motion abnormalities of the left ventricle (LV) assessed by echocardiography and symptomology (syncope or cardiac arrest) in patients with LQTS [63–65]. Using an M-mode technique, these investigators demonstrated the presence of a very slow contraction phase before rapid relaxation that manifested as either a plateau or as a double-peaked contraction in over half of the studied patients with LQTS. These contraction abnormalities, in particular the double-peaked morphology, were associated strongly with a clinical history of syncope or cardiac arrest [63]. Subsequently, calcium channel blockade by verapamil was illustrated to normalize even the most severe abnormality patterns, including those with a double-peak morphology [64]. The presence of a slow contraction phase before rapid relaxation in LQTS patients was confirmed in 1998 by Nakayama [66]. In 2003, Savoye and colleagues utilized TDI to assess wall motion and myocardial velocities in ten patients with ‘mild’ LQTS and 14 control subjects and observed similar results as Savoye and colleagues [67]. In 2009, Haugaa and colleagues investigated whether prolonged and dispersed myocardial contraction duration assessed by TDI may serve as a risk marker for cardiac events in patients with LQTS [68]. Here, 73 patients with genetically confirmed LQTS (9 with JLNS, 33 symptomatic subjects with single mutations, and 31 asymptomatic subjects with single mutations) and 20 healthy control subjects were studied. Myocardial contraction was significantly prolonged in each LQTS patient group compared to the controls. Furthermore, contraction duration and dispersion of contraction was significantly longer in symptomatic patients (0.46 ± 0.06 s; 0.048 ± 0.018 s) compared to asymptomatic LQTS patients (0.40 ± 0.06 s, P = 0.001; 0.031 ± 0.019 s, P = 0.001) [68]. In fact, while a QTc ≥ 0.46 s showed a sensitivity of 70 % (95 % CI 67–88) and specificity of 50 % (95 %, CI 40–61) in identifying symptomatic individuals, contraction duration with an optimal cut off value of 0.44 s had a sensitivity of 79 % (95 % CI 68–87) and specificity of 74 % (95 % CI 62–83). Thus, assessment of myocardial duration may add an important clinical parameter for risk stratification in LQTS mutation carriers, in particular for those subjects with a normal or mildly prolonged QTc [68].
Molecular Genetic Testing
The discipline of cardiac channelopathies was birthed publicly on March 10, 1995, with tandem publications by the Keating laboratory in Cell that revealed LQTS to be a disease of the cardiac channels [69, 70]. These foundational discoveries permitted research-based genetic testing for LQTS over the ensuing decade. In May 2004, akin to the evolution from research testing to clinical testing for its sister channelopathy, cystic fibrosis, LQTS genetic testing completed its maturation into a clinical diagnostic genetic test involving a comprehensive open reading frame analysis of the translated exons for the genes associated with LQT1, LQT2, LQT3, LQT5, and LQT6. In the United States, Transgenomic (www.familion.com), GeneDx (www.genedx.com), and Correlagen Diagnostics (www.correlagen.com) currently represent three CLIA-approved clinical laboratories that offer fee-based clinical genetic testing for LQTS, with LQTS gene testing panels that now include LQT1–LQT13.
In 2011, two consensus documents, the Heart Rhythm Society (HRS)/European Heart Rhythm Association (EHRA) Expert Consensus Statement [71] and the Canadian Cardiovascular Society (CCS)/Canadian Heart Rhythm Society (CHRS) joint position paper [72] were published on the use of genetic testing in the clinical evaluation of cardiac channelopathies and cardiomyopathies. Accordingly, “comprehensive or LQT1-3 targeted genetic testing is recommended for any patient in whom a cardiologist has established a strong clinical index of suspicion for LQTS based on examination of the patient’s clinical history, family history, and expressed electrocardiographic phenotype.” Genetic testing is also recommended for any asymptomatic patient with QT prolongation (QTc > 480 ms, prepubertal; QTc > 500 ms, adult) in the absence of other clinical conditions that might prolong the QT interval (i.e. electrolyte abnormalities, hypertrophy, bundle branch block, etc.). Mutation-specific genetic testing is recommended for family members following the identification of a LQTS-causing mutation in the index case. For any asymptomatic patient with otherwise idiopathic “borderline” QTc values of >460 ms (pubertal) or >480 ms (adult), LQTS genetic testing may be considered [71].
When there is no doubt about the clinical diagnosis, the genetic test will be positive about 75 % of the time [43, 73, 74]. Identification of the specific LQTS-associated mutation then assists with genotype-guided therapy and provides the gold standard diagnostic marker for the unambiguous genetic classification of all relatives. An estimated 5–10 % of LQTS probands have spontaneous germline mutations (sporadic) and 5–10 % of patients who are genotype positive have >1 mutation [73–77]. Like patients with JLNS (autosomal recessive LQTS plus deafness), this subset of patients with multiple mutations tend to have a more severe phenotype [76]. These observations underscore the critical importance for comprehensive genetic testing rather than a “find-and-stop” approach.
To be sure, a negative genetic test MUST NOT result in removal of the diagnosis in a patient with a high probability/definite “Schwartz score”. Remember, 20–25 % of patients with clinically definite LQTS will have a negative genetic test. On the other hand, there appears to be an abundance of patients for whom the diagnosis of LQTS may have been assigned prematurely based upon rather soft criteria. In this setting, a negative genetic test result which objectively and unemotionally “RULES OUT” at least 75 % of LQTS may be a helpful piece of data for a specialist to help the patient/family move away from the diagnosis. Among patients referred for LQTS genetic testing despite an intermediate probability “Schwartz score”, 44 % had a positive genetic test enabling their status to be elevated from diagnostic ambiguity (borderline LQTS) to diagnostic certainty [43]. Finally, the physician must be aware that the LQTS genetic test has an estimated 5 % false positive rate [78–80]. That is, approximately 5 % of healthy individuals are positive for a rare genetic variant in these LQTS-susceptibility genes. These rare variants tend to localize to the N- and C-termini of the LQT1- and LQT2-associated potassium channels and to the N-terminus, C-terminus, and cytoplasmic inter-domain linkers of the SCN5A-encoded sodium channel (LQT3) [80, 81].
Given the severe consequences surrounding the misdiagnosis and mismanagement of patients with these potentially lethal cardiac disorders, the clinical evaluation and management of a patient and family suspected of having LQTS should be performed under the supervision of a pediatric or adult cardiologist with experience and expertise in heritable cardiac channelopathies [82]. Because of the issues associated with incomplete penetrance and variable expressivity, the genetic test result must be interpreted cautiously and incorporated into the overall diagnostic evaluation for this disorder. Counter to current widespread thought, genetic testing in general should not be considered binary but rather probabilistic in nature. In fact, even when a genetic variant has been published previously as a putative pathogenic mutation, assignment of a specific genetic variant as a true disease-causing mutation still requires some vigilant scrutiny.
Establishing the “background genetic noise rate” for the three most common LQTS genes has enabled a case: control mutational analysis of the properties and localization of case-associated mutations compared to the compendium of presumably innocuous variants [83]. Algorithms based on mutation location, species conservation, and the biophysical nature of the amino acid substitution may assist in distinguishing pathogenic mutations from otherwise rare variants of uncertain significance (VUS) and perhaps allow for the assignment of estimated predictive values for the probability of pathogenicity of each novel mutation identified within a specific gene [80]. The probabilistic rather than binary nature of genetic testing for LQTS is depicted in Fig. 27.6 showing that rare “radical” mutations (i.e. insertion/deletion, frame-shift, nonsense, or splice-site mutations which represent ∼20 % of the LQTS spectrum of mutations) are high probability LQTS-associated mutations whereas the probability of pathogenicity for the most common mutation type, missense mutations (i.e. single amino acid substitutions), is strongly location dependent. For example, missense mutations localizing to the transmembrane spanning/pore domains of the LQT1- and LQT2-associated potassium channels are high probability disease mutations whereas a similarly rare missense mutation that localizes to the domain I-II linker of the Nav1.5 sodium channel is indeterminate, a variant of uncertain significance (VUS). Without co-segregation or functional data, such a mutation has a point estimate for probability of pathogenicity of <50 % [80].
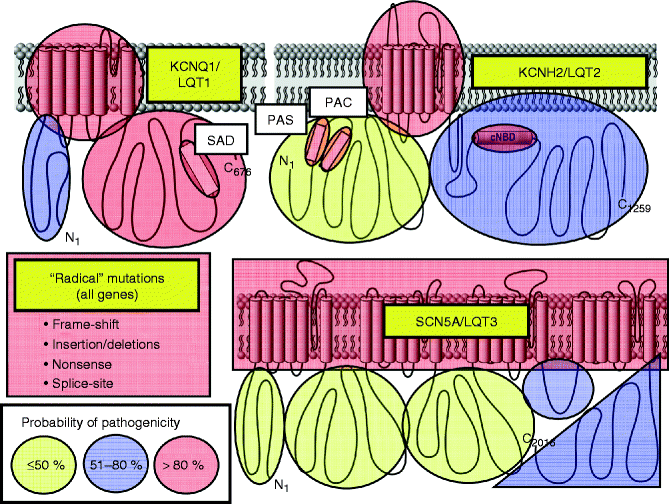
Figure 27–6.
Probabilistic nature of LQTS genetic testing. Depicted are the three major ion channels causative for LQTS with areas of probability of pathogenicity shown for mutations localizing to these respective areas. While “radical” mutations have a >90 % probability of being a true pathogenic mutation, the level of probability for missense mutations vary depending on their location for each channel protein. Missense mutations residing in red shaded areas have a high probability (>80 %) of being pathogenic, those in blue are possibly (51–80 %) pathogenic, and those in yellow shaded areas truly represent variants of uncertain significance (VUS, ≤50 % probability) clinically [80] (Adapted from Tester and Ackerman [19] with permission from Elsevier)
Thus, it is critical that the physician carefully synthesize together all components of the diagnostic evaluation. This meticulous, comprehensive approach is vital to ensure that the wisest recommendations are rendered.
Management
When LQTS was estimated to affect 1:20,000 persons, the disease was viewed as a “death sentence” with an untreated annual mortality of 5–10 %. Akin to the “tip of the iceberg” phenomenon, LQTS has become much more common (1 in 2,000) and much less severe (overall annual mortality ∼1 %) over the last half century since its original description in 1957.
Risk Stratification
The issue of risk stratification in LQTS is clinically important. The phenotypic expression of LQTS varies profoundly from asymptomatic longevity to premature sudden cardiac death despite medical therapy. The great challenge is to try to discern which of these divergent outcomes is most likely in each of our patients. Occurrence of a LQTS-related cardiac event like syncope before 5 years of age suggests a serious LQTS phenotype and syncope occurring in the first year of life is associated with an extremely poor prognosis [84].
Overall among LQTS patients, the risk of cardiac events is higher in males until puberty and higher in females during adulthood [16]. Events tend to occur earlier in males than females, and males who are still asymptomatic at age 20 years may be considered at lower risk (lowest in LQT1) for manifesting cardiac events [16, 85]. Even if event free for the first 20 years of life, females continue to have discernible risk for cardiac events in adulthood and may be at increased vulnerability to an arrhythmic event during the postpartum period [17].
Among the known LQTS genotypes, patients with LQT1 are “frequent fainters” while patients with LQT3 have the highest lethality rate per cardiac event [18, 30]. The frequency of cardiac events including syncope, aborted cardiac arrest and sudden death was highest in LQT1 (60 % of patients), then LQT2 (40 %) and lowest in LQT3 (18 %) [30]. Since the rate of death was the same in each of these three genotypes, the percentage of lethal events was highest in LQT3 patients. JLNS is associated with very early clinical manifestations and a poorer prognosis than autosomal dominant Romano-Ward LQTS [9, 86]. Patients with JLNS due to compound homozygous/heterozygous mutations involving KCNQ1 (JLN1) have a significantly greater risk than patients with JLN2 due to mutations involving KCNE1 [86]. Patients with the very rare form of LQTS with syndactyly, now known as type 1 Timothy syndrome secondary to mutations in the L-type calcium channel, have a very poor prognosis [87, 88].
Intra-genotype risk stratification and in rare instances, mutation-specific risk stratification, has also been realized for the two most common subtypes of LQTS based upon mutation type, mutation location, and cellular function [89–94]. In general, LQT1 patients with KCNQ1 (Kv7.1) missense mutations localizing to the transmembrane-spanning domains clinically have a two-fold greater risk of a LQT1-triggered cardiac event than LQT1 patients with C-terminal region localizing mutations. Trumping location, patients with mutations resulting in a greater degree of Kv7.1 loss-of-function at the cellular in vitro level (i.e. dominant negative) have a two-fold greater clinical risk than those mutations that damage the biology of the Kv7.1 channel less severely (haploinsufficiency).
Besides the observations that transmembrane localizing and dominant negative functioning mutations represent higher risk LQT1 mutations than C-terminal localizing and haploinsufficient ones, Crotti and colleagues illustrated the potential for mutation-specific risk stratification by providing a phenotypic clinical severity data from a large group of individuals (n = 78) from 21 families and 8 countries (different parts of the world and ethnic background) and a South African founder population that hosted a specific LQT1 mutation (A341V-KCNQ1) compared to a group of 205 patients with non-A341V LQT1[95]. Despite the in vitro mild dominant negative functional effect of this Kv7.1 transmembrane region mutation, the LQT1 A341V population had a significantly greater percentage of symptomatic patients, manifesting with their first cardiac event at an earlier age, higher incidence of life-threatening arrhythmias, more prolonged mean QTc, lower frequency of asymptomatic (“silent”) mutation positive subjects, and twice the proportion of patients with a QTc ≥ 500 ms compared to the non-A341V group [95].
Akin to molecular risk stratification in LQT1, patients with LQT2 secondary to KCNH2 (Kv11.1) pore region mutations have a longer QTc, a more severe clinical manifestation of the disorder, and experience significantly more arrhythmia-related cardiac events occurring at a younger age than those LQT2 patients with non-pore mutations in Kv11.1 [89]. Similarly, in a Japanese cohort of LQT2 patients, those with pore mutations had a longer QTc, and while not significant among probands, non-probands with pore mutations experienced their first cardiac event at an earlier age than those with a non-pore mutation [93]. In addition, LQT2 patients with mutations involving the transmembrane pore region have the greatest risk for cardiac events, those with frame-shift/nonsense mutations in any region have an intermediate risk, and those with missense mutations in the C-terminus have the lowest risk for cardiac events [94]. Adding to the traditional clinical risk factors, molecular location and cellular function are independent risk factors used in the evaluation of patients with LQTS [92].
Now, in 2012, a recent discovery has elucidated a potent genetic determinant that underlies penetrance and expressivity in LQT1 [96]. Here, a specific set of KCNQ1 single nucleotide polymorphisms (SNP, rs2519184, rs8234, and rs10798) residing in the 3′untranslated region (UTR) of the gene modify LQT1 disease severity in an allele-specific manner, by suppressing the allele on which they reside through microRNA-mediated repression of transcription and translation [96]. When these SNPs reside on the same allele that contains the LQT1 mutation, the expressivity is attenuated (less mutant protein is made by the cardiomyocyte, thus proportionally more normal protein). However, when they reside on the normal allele (allele not containing the mutation), the disease phenotype is more severe (less normal protein is made by the cardiomyocyte, thus proportionally more mutant protein) [96]. If validated, this observation may cause a paradigm shift in our understanding of autosomal dominant genetic diseases like LQTS with the discovery that one of the single greatest determinants of disease severity depends on what was inherited from the normal parent.
A history of recurrent cardiac arrest increases the probability of SCD at follow-up [97]. Except resuscitated SCA, syncope is one of the strongest sudden death warning signs [14]. This observation now presses to the level of critical importance the proper distinction between vasovagal syncope and torsadogenic syncope less the trend towards early ICD therapy be accelerated even further. A negative family history for SCD cannot be regarded as a predictor of favorable outcomes. On the other hand, a family history of SCA may not indicate increase risk for the affected relatives left behind.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


