CONGENITAL HEART DISEASE
CHAPTER

Congenital Heart Disease in the Adult
Adults with congenital heart disease (CHD) are a rapidly growing population of patients owing to advances in the diagnosis and treatment of children with CHD. Most children with CHD are now expected to survive to adulthood either with or without the aid of surgical correction or palliation. According to recent estimates, there are now nearly a million adults with CHD, and these numbers should continue to rise with further advancements in diagnosis and treatment. Although ideally served by cardiologists with advanced training in adult CHD, most of these patients receive the majority of their care from primary care physicians and general cardiologists, even though few cardiology training programs have a formalized adult CHD curriculum. Being aware of the often unique clinical presentations, and having a general understanding of the anatomy and the pathophysiologic consequences of congenital disease is vital to facilitating the timing of percutaneous, electrophysiologic, and surgical interventions.
GENERAL CONCEPTS
An organized approach to diagnosis and management is especially important in patients with CHD, and the critical first step is gathering historical data. Reviewing the pediatric and operative records, if available, is essential in understanding the complexities of the cardiac and vascular anatomy and to define the outcomes of previous diagnostic studies and surgeries. Surgical procedures have changed considerably over the last several decades, and anatomic presumptions based on current practice may not apply.
Certain signs and symptoms should prompt an extensive evaluation of adults with CHD, particularly syncope and progressive exertional dyspnea. Arrhythmias are not uncommon in adults with CHD and often originate near the myocardial scars of previous surgeries. The most common of tachycardia seen is macroreentry within the atrial muscle. Supraventricular arrhythmias, such as atrial flutter or fibrillation, are often poorly tolerated due to a dependence on atrial mechanical function. Ventricular arrhythmias, typically microreentrant ventricular tachycardia, which can result in sudden death, may develop in adult patients with CHD as a late complication of prior ventriculotomy and patching of a ventricular septal defect (VSD). The incidence of ventricular arrhythmias in adults with corrected tetralogy of Fallot (TOF) is estimated between 0.5% and 6%, with independent risk variables that include a widened QRS interval (>180 milliseconds) on a surface electrocardiogram (ECG), significant right ventricular dilation, and older age at time of surgical repair.
Hemodynamic derangements can be quite subtle, such as pulmonic regurgitation following a patch outflow repair of TOF. Since pulmonary regurgitation has a low-pressure gradient, it can be missed during auscultation and routine echocardiography and can eventually result in right ventricle (RV) enlargement and increased risk of sudden death.
Diagnostic imaging is a critical adjunct, and less invasive modalities such as echocardiography are an important first step (see Chapter 24). Limitations of echocardiography include difficult windows due to excessive scar tissue from previous surgeries, concomitant lung disease, and obesity. Subsequent computerized tomography (CT) scanning or magnetic resonance imaging (MRI) may add substantially to the anatomic description, especially in patients with unclear great vessel or pulmonary vascular anatomy. The use of MRI has expanded with more widely available scanners and simplified scanning protocols. It is important to remember, however, that CT scanning is complicated by the need for intravenous contrast and exposure to radiation, and MRI is generally not compatible with current implantable cardiac devices.
Diagnostic cardiac catheterization, though generally performed later in the diagnostic workup of CHD patients than in the past, remains the gold standard for pressure measurement, cardiac output calculation, and vascular resistance determination. The relative size of shunts lesions can be assessed using oximetry, and the hemodynamic consequences of additional blood flow can be assessed. Most importantly, cardiac catheterization affords the opportunity to intervene and palliate or repair anatomic defects or to clarify the suitability of further surgical intervention.
Anatomic shunting can be quantified in the catheterization laboratory by examining the blood oxygen saturations in the respective chambers. The mixed venous (MV) saturation is the saturation of blood returning to the right atrium (RA) with contributions from the inferior vena cava (IVC), superior vena cava (SVC), and coronary sinus (CS). IVC saturation is normally higher than the SVC due to high renal blood flow and less oxygen extraction by the kidney. The CS saturation is very low, but its volume of contribution is negligible and usually ignored. To normalize the MV saturation, three times the SVC saturation is added to the IVC saturation and the sum divided by 4.
Because so much mixing of blood with differing saturations occurs in the RA, an 11% increase in oxygen step-up (saturation increase from a chamber to its successive chamber) is required to diagnose a shunt lesion between the SVC and the RA. A 7% increase is necessary to detect a shunt between the RA and the RV and a 5% increase to detect a shunt between the RV and the pulmonary artery (PA). A quick and simple measure of the overall size of a left-to-right shunt ratio can be obtained by using the formula: (aortic saturation-MV saturation)/(PV saturation-PA saturation). The PV saturation can be assumed to be 97% if not directly measured.
In general, a “significant shunt” is present when the shunt ratio is ≥1.5:1.0. This simplified definition may not apply to older adults, however. As pulmonary hypertension develops and RV compliance falls, a left-to-right shunt that was 3:1 for 30 years may become <1.5:1 due to the gradual reversing of the shunt. In fact, the left-to-right shunt may totally reverse at some point and result in arterial desaturation, the so-called Eisenmenger syndrome. The significance of a shunt in the adult must, therefore, be examined in the context of the other hemodynamics, chamber sizes, and the history of the defect over time.
Pulmonary hypertension is a frequent complication of certain CHDs. It can be secondary to pulmonary venous hypertension from elevated left-sided filling pressures, or the result of systemic-to-PA shunting. For unclear reasons, shunts proximal to the tricuspid valve (atrial septal defects [ASDs] or partial anomalous pulmonary venous return) infrequently result in pulmonary hypertension (~15% of cases) despite high pulmonary blood flow. The development of pulmonary hypertension from shunts distal to the tricuspid valve, however, is very dependent on pulmonary blood flow. For example, a large unrestricted VSD may not result in pulmonary hypertension if the pulmonary circuit is protected by concomitant pulmonary valvular or subvalvular obstruction.
To help differentiate the cause of pulmonary hypertension, the pulmonary vascular resistance should be determined: (mean PA pressure-mean pulmonary capillary wedge pressure [mm Hg])/(pulmonary blood flow [liters per minute]). Higher resistances (>7 Wood units or a ratio of the pulmonary-to-systemic vascular resistance of >0.5) have been associated with considerably higher perioperative mortality. In addition, assessment of pulmonary vascular reactivity with endothelium-dependent vasodilators, such as inhaled nitric oxide or intravenous adenosine, may provide additional prognostic information in these patients by confirming whether any of the observed pulmonary hypertension has a vasoconstrictor component. In patients with shunt lesions and pulmonary arterial hypertension (mean PA pressure ≥25 mm and mean pulmonary capillary wedge pressure ≤15 mm Hg), growing evidence supports the use of selective pulmonary vasodilator therapy (such as endothelin blockers and phosphodiesterase-5 inhibitors) to improve exercise capacity and reduce symptoms. No patients with CHD should be started on these medications, however, without first undergoing thorough hemodynamic assessment in the catheterization laboratory.
TYPES OF CONGENITAL LESIONS
Congenital heart lesions can be divided into three general categories (by descending incidence): simple shunt lesions, obstructive lesions, and complex lesions—acyanotic and cyanotic. The most frequently encountered abnormalities in these categories are mentioned below.
Shunt Lesions
Intracardiac shunts are the most common form of congenital heart lesion and are frequently diagnosed in otherwise healthy adults. They are associated with increased pulmonary blood flow, which can lead to right heart chamber enlargement and arrhythmias, as well as pulmonary hypertension. The surgical correction of many of these lesions has been determined to be safe and efficacious. Recently, percutaneous device closures have been increasingly utilized in order to avoid the morbidity and mortality of surgery. There are three main types of shunt lesions to be aware of: ASD, VSD, and patent ductus arteriosus (PDA). All these lesions demonstrate left-to-right shunting under normal physiologic conditions.
Atrial Septal Defect
The ASD is the most common congenital heart defect encountered in adults (excluding mitral valve prolapse and bicuspid aortic valve), accounting for up to 15% of all adult CHD. It results from the failure of proper embryologic development of the atrial septum. Almost a third of patients with ASD will have associated additional malformations such as pulmonary stenosis, VSD, mitral valve prolapse, subaortic stenosis, aortic coarctation, and anomalous pulmonary venous drainage.
There are many different types of ASD (see Chapter 24), the most common of which (75% of the cases) is the secundum ASD, in which the defect lies in the middle of the atrial septum. The secundum ASD is often mistaken for other abnormalities or overlooked because the symptoms associated with it, typically fatigue, palpitations, and breathlessness, can be subtle and nonspecific. Other less common variations of ASD include the sinus venosus ASD in which there is abnormal fusion of the vena cava (superior or inferior) to the left atrium. This defect is almost always associated with partial anomalous return of the pulmonary veins (right superior or both right pulmonary veins draining into the SVC or RV). Because of its location, this defect can be missed on transthoracic echocardiography and usually requires either transesophageal echo or advanced radiographic imaging to make the diagnosis. The primum ASD involves the lower portion of the atrial septum and typically affects the ventricular septum as well (the so-called AV canal defect). Both AV valves are structurally abnormal and the mitral valve is typically cleft. This defect is commonly seen in patients with trisomy 21 (Down syndrome). The least common ASD, the CS septal defect, involves unroofing of the CS, which results in shunting from the left to the RA. Commonly, a persistent left SVC or an abnormal pulmonary venous drainage accompanies CS ASD. Important differences in the clinical findings among the various types of ASD are listed in Table 23.1.
TABLE
23.1 Unique Features of the ASDs
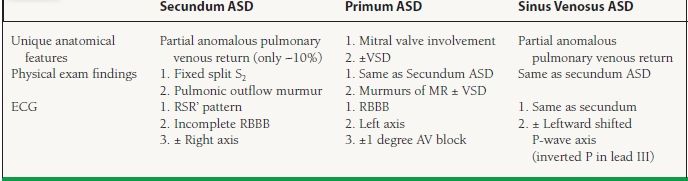
ASD should be suspected whenever right heart enlargement is present without an alternative explanation. Physical examination findings, such as a fixed split second heart sound (due to loss of differential effects on right- and left- sided filling pressures from a drop in intrathoracic pressure that normally occurs during inspiration) and a pulmonic outflow murmur (the result of increased pulmonary blood volume from shunting), can also be overlooked. The flow of blood across the defect (shunt) is determined by the size of the defect and the compliance of the atria. Occasionally, patients can present late in life with ASD-related symptoms when the left atrial pressure rises because of a stiff left ventricle and diastolic dysfunction (usually the result of long-standing hypertension or coronary artery disease). On electrocardiography, an incomplete right bundle branch block, right-axis deviation, abnormal P-wave axis, and right atrial enlargement are commonly seen. Due to anatomic position of the conduction bundles, a superior left axis is usually noted in primum ASD. On chest x-ray prominent pulmonary arteries, right atrial and ventricular enlargement and pulmonary plethora can be seen.
The larger the left-to-right shunt in patients with ASD, the greater is the risk for long-term complications such as atrial fibrillation (typically occurring in the fifth decade) and pulmonary hypertension. The latter condition affects up to 5% to 10% of adults with ASD, and if left uncorrected can result in Eisenmenger syndrome. In Eisenmenger syndrome, the pulmonary vascular resistance increases to the point that shunting is reversed (becoming right to left) and systemic oxygenation decreases. Patients with this complication will not improve their oxygen saturation when oxygen is administered to them (the telltale sign of a right-to-left shunt). Multiple complications eventually ensue, and until recently, this condition was considered irreversible. Another condition associated with ASD is stroke, which presumably results from paradoxical embolization (blood clots forming in the extremities and reaching the cerebral circulation by passing through the ASD).
The guideline-based indication to repair an ASD is right heart enlargement from volume overload resulting from the ASD, regardless of whether the patient is symptomatic or asymptomatic. At this time, only the secundum ASD has been successfully occluded through percutaneous methods. All other types of ASD require surgical closure.
Repair of an ASD may also be reasonable in the context of paradoxical embolism or documented platypneaorthodeoxia and should be considered in the presence of a hemodynamically significant net left-to-right shunt and PA pressure or pulmonary vascular resistance <2/3 systemic levels or if the pulmonary hypertension is vasoreactive. The timing of closure of an ASD appears important. Closure after the age of 40 is associated with an increased incidence of arrhythmias (i.e., atrial fibrillation) compared with closure before age 40. Epidemiologic evidence also suggests that long-term survival is worse with unrepaired defects, but the difference is lost as the patient is older at the time of repair and outcomes may be worsened with repair after pulmonary hypertension has become well established.
VSD is the most common congenital heart defect seen in children. Defects can occur at various locations in the septum but most commonly occur in either the membranous or the muscular portions (see Chapter 24). Perimembranous VSD is the most common (80% of the cases), occurring in the membranous septum and adjacent to the septal tricuspid valve leaflet. Small defects, typically muscular VSDs, often close spontaneously during childhood. One type of defect, the so-called outflow (or supracristal VSD), can be occluded by one of the aortic leaflets prolapsing into it. This can result in the development of rather significant and progressive aortic regurgitation. VSDs are often isolated lesions but are also common defects in complex abnormalities such as TOF and congenitally corrected transposition of the great vessels.
Small VSDs produce a very loud systolic murmur and frequently a palpable thrill at the left sternal border. Patients with small defects are asymptomatic and require regular follow-up without a need for intervention. Larger defects have less conspicuous murmurs.
Clinical presentation in adulthood depends on the defect size and the pulmonary vascular resistance. Left-to-right shunting across the defect can lead to left ventricular volume overload and pulmonary hypertension. Large defects are more likely to present in childhood with symptoms of heart failure. VSD is also the most frequent cause of Eisenmenger syndrome, with shunt reversal to right to left. The unrepaired adult may present with symptoms of heart failure, exercise intolerance, infective endocarditis (IE), pulmonary hypertension, and cyanosis or arrhythmias.
On electrocardiography, patients with large VSD and significant pulmonary hypertension will have isolated right ventricular or biventricular hypertrophy. The chest x-ray typically is normal in patients with small VSD; however, in those with large left-to-right shunt, left atrial and left ventricular enlargement as well as increased pulmonary vascular markings may be noted.
Cardiac catheterization is helpful in assessing the operability of an adult patient with VSD and pulmonary hypertension, including quantification of shunting and assessment of pulmonary pressures, pulmonary vascular resistance, and pulmonary vasoreactivity.
VSD closure is indicated with evidence of left ventricular volume overload, large pulmonary-to-systemic flow (Qp/Qs >2), or in patients with history of IE. Closure of a VSD may also be reasonable in the setting of a Qp/Qs >1.5 with left ventricular systolic or diastolic dysfunction or with PA pressure and pulmonary vascular resistance <2/3 systemic levels. VSD closure is usually accomplished surgically, although some muscular VSD may be amenable to catheter device closure.
Patent Ductus Arteriosus
PDA, the second most common congenital heart defect seen in adults (~10% to 15% of all CHD in adults), is a persistent communication between the descending aorta and the left PA at the level of the left subclavian artery. It has been associated with maternal rubella. PDA is present as an isolated lesion in most adults, unlike in children where it is frequently seen with more complex heart defects. Patients with PDA have a continuous murmur (systole and diastole) that is often described as a “machinery murmur,” heard best under the left clavicle and accompanied by a widened pulse pressure. Like in VSD, patients with a large, uncorrected PDA can present with dyspnea and easy fatigability as well as Eisenmenger physiology with differential cyanosis and clubbing.
The ECG of adults with PDA may be normal or show left atrial enlargement, left ventricular enlargement, or right ventricular hypertrophy (in setting of pulmonary hypertension). The chest x-ray may show cardiomegaly and increased pulmonary venous markings depending on the size of the shunt. The need for closure of a PDA in adults is uncommon. Defects can be ligated surgically or closed percutaneously (device closure or coils) depending on size.
Stenotic Lesions
Pulmonary Stenosis
Pulmonic stenosis (PS) is the most common congenital valve lesion to necessitate therapy in adults. It is occasionally associated with Noonan syndrome, in which the valve is usually dysplastic. Gradients across the pulmonary outflow tract can involve the valvular level, but may also involve the infundibulum (right ventricular outflow tract) and/or the peripheral pulmonary arteries. Careful tracking of the gradient is critical for decision making.
Patients may present with an asymptomatic systolic murmur or with exercise intolerance. Findings on cardiac examination depend on the severity of the PS, pathology of the valve, and any associated lesions. Patients may have elevated jugular venous pressure with a prominent “A” wave, a right ventricular heave, a systolic ejection murmur, an ejection click that decreases with inspiration, a wide splitting of S2, and/or a reduced or an absent P2.
On electrocardiography of PS, right atrial enlargement, right-axis deviation, and right ventricular hypertrophy may be noted. Chest radiography demonstrates right atrial enlargement, dilation of the PA, and occasionally vascular fullness in the left base greater than the right base (Chen sign) due to preferential blood flow to the left lung.
Generally, the diagnosis of PS is accomplished with echocardiography. An intervention is felt warranted when the peak instantaneous transvalvular gradient exceeds 60 mm Hg in an asymptomatic patient (mean Doppler gradient >40 mm Hg) or in a symptomatic patient with a peak gradient >50 mm Hg (mean Doppler gradient >30 mm Hg), though patients with lesser gradients may benefit if it can be clearly shown that exertional symptoms (typically exertional dyspnea) accompany elevated gradients during provocation. Interventions on PS can include percutaneous balloon valvuloplasty, surgical pulmonary valvotomy, or valve replacement with the balloon procedure generally favored.
Aortic coarctation (CoA) is a common congenital heart defect, accounting for approximately 8% of all congenital defects. It likely results from extraneous ductal tissue that contracts following birth. It is associated with Turner syndrome. Anatomically it can occur before, at the level of or after the ductus arteriosus, though adults with previously undiagnosed CoA will almost always have postductal lesions. The most common presentation in adults is the fortuitous discovery during secondary workup for systemic hypertension. Lower extremity and renal hypoperfusion leads to a hyperrenin state that may not abate even after coarctation repair. In most patients, there is upper-extremity hypertension and the development of collateral vessels around the coarctation to the lower extremity. These collateral channels result in a continuous murmur heard over the back, and involvement of the intercostal arteries leads to the familiar rib notching noted on chest x-rays. Extensive collateral vessels may mask the severity of the obstruction by reducing the gradient across the coarctation. Associated cardiac defects include bicuspid aortic valve (present in up to 85% of cases), subaortic stenosis, VSD, mitral valve abnormalities, aortic aneurysms as well as cerebral aneurysms in the circle of Willis (seen in up to 10% of patients and which can lead to CNS bleeding).
Adults with CoA may present with hypertension and discrepant upper- and lower-extremity pulses. They may complain of claudication, leg fatigue, or exertional headaches. Accelerated coronary artery disease, stroke, aortic dissection, and congestive heart failure are common complications in the unoperated patient or those intervened on in childhood. On examination, hypertension is present in the right arm relative to the lower extremity. A radial-femoral pulse delay may be noted. There may be an ejection click with a systolic murmur in the setting of a bicuspid aortic valve. Continuous murmur in the parasternal areas and around the left scapula may be heard due to the flow through collateral vessels.
Electrocardiographic findings in aortic coarctation may include left ventricular hypertrophy with associated repolarization abnormalities. On chest x-ray, rib notching due to collateral vessels may be noted as well as dilation of the proximal aorta and a “3 sign” due to indentation at the coarctation site.
Echocardiography with a focus on the descending aorta is an excellent noninvasive test to make the clinical diagnosis in patients with suspicious clinical findings. Cardiac MRI and CT scanning can identify the precise location and anatomy of the coarctation and assess the entire aorta and the collateral vessels. Cardiac catheterization is indicated to assess for concomitant coronary artery disease when surgery is planned as well as when catheter-based interventions are contemplated.
Repair of coarctation of the aorta may be accomplished via percutaneous catheter intervention or surgically. The main indication for intervention of CoA is a peak-to-peak coarctation gradient >20 mm Hg. Intervention may also be sought with lower gradients in the setting of significant aortic narrowing on anatomic imaging with radiologic evidence of extensive collateral flow.
Surgery was previously the mainstay in the approach to native CoA, with available options including resection and end-to-end anastomosis, subclavian flap, prosthetic patch aortoplasty, and interposition (tube bypass) grafting. Angioplasty and stenting is now considered the procedure of choice in patients with recoarctation following surgery and is experiencing a greatly expanding role in treatment of primary CoA.
Complex Lesions (Acyanotic)
Transposition of the Great Arteries
Transposition of the great arteries (TGA) refers to an abnormality in the embryologic separation of the great vessels, which results in the aorta emanating from the RV and the PA coming off the left ventricle. There are two varieties that are most commonly seen in adults: D-TGA and L-TGA.
In D-TGA, the great arteries arise from the wrong ventricle; as such surgical intervention is required during childhood. The surgery may include an atrial baffle (a Senning or Mustard procedure) where blood is baffled from the vena cavae to the left atrium and from the pulmonary veins to the RA. The primary long-term concern in these patients is that the RV is ill-prepared to serve as a systemic ventricle. It can weaken and fail over time (usually when the patient enters their 30s and 40s), and these patients typically also develop significant systemic atrioventricular valve (SAVV) (tricuspid valve in the mitral position) regurgitation. Other common complications include baffle obstruction, baffle leak, pulmonary venous obstruction, and conduction disturbances and arrhythmias.
Patients with D-TGA can also be surgically corrected during childhood with an arterial switch (Jatene procedure), where the ascending aorta and the main PA are transected and reattached to the opposite root with transplantation of the coronary arteries into the “neoaorta.” Long-term concerns after this operation include coronary insufficiency with myocardial ischemia, stenosis at the anastomotic sites, ventricular dysfunction and arrhythmias, and aortic and pulmonary regurgitation.
The ECG may be normal in D-TGA (post arterial switch) or may reveal right-axis deviation and right ventricular hypertrophy (postatrial baffle). On chest radiography, because of the parallel relationship of the great vessels, a narrow mediastinal shadow is common. Ventricular size and pulmonary markings vary depending on the patient’s clinical status.
L-TGA is the so-called congenitally repaired lesion and consists of atrioventricular and ventricular-arterial discordance. This variation results in a circulation where the circulation goes from vena cavae to RA to left ventricle to PA to pulmonary veins to left atrium to RV to aorta. Associated anomalies are common in L-TGA and include VSD, PS, abnormalities of the SAVV, and conduction abnormalities, with complete heart block occurring at a rate of approximately 2% per year. Many patients with L-TGA may be asymptomatic and escape diagnosis until adulthood. They may present with heart failure due to significant SAVV regurgitation or systemic ventricular dysfunction, arrhythmias, or complete heart block.
On electrocardiography, PR prolongation may be noted and there may be complete heart block. Because of the inversion of the left and right bundle branches, a pattern of inferior infarction may be seen. On chest radiography, the vascular pedicle may be abnormal or narrow and the ventricular silhouette has a “humped” appearance. Cardiomegaly may be noted as well as dextrocardia, which also occurs with D-TGA.
Again, in L-TGA, the problem remains a RV pumping into the systemic circulation. Surgical repair for important SAVV regurgitation should be undertaken before the systemic ventricular function deteriorates (ejection fraction <45%). Patients with systemic ventricular dysfunction may benefit from therapy with beta-blockers and afterload reducers such as ACE inhibitors or angiotensin II receptor blockers. Cardiac transplantation is often considered in the patients with severe systemic ventricular dysfunction refractory to medical therapy.
Complex Lesions (Cyanotic)
Tetralogy of Fallot
TOF, a so-called conotruncal abnormality, is the constellation of four findings: an aorta that overrides the right ventricular outflow tract, right ventricular outflow obstruction, a large subaortic VSD, and hypertrophy of the RV The frequent coexistence of an ASD can make for a “pentalogy.” Other associations with TOF include a right aortic arch in a fourth of the patients as well as anomalous left anterior descending coronary artery arising from the right coronary artery, passing anterior to the right ventricular outflow tract. Occasionally, unrepaired patients with TOF can present in adulthood owing to a remarkable balance between the pulmonic obstruction and the VSD, which limits cyanosis.
Early palliation with a systemic to arterial shunt (i.e., Blalock–Taussig, which connects the subclavian artery and the PA) facilitates growth of the pulmonary arteries and is a precursor to definitive surgical repair in the young child. Definitive repair comprises closure of the VSD, as well as relief of the right ventricular outflow tract obstruction, which may include simple resection of infundibular stenosis, patch augmentation of the right ventricular outflow tract that may disrupt the pulmonary valve or pulmonary valvotomy resulting in significant pulmonic regurgitation. Though tolerated for several years, the RV eventually succumbs to volume overload and progressively increases in size. Surgical repair of this condition involves implanting a pulmonary homograft or a pulmonary valve bioprosthesis, the latter of which may now be able to be implanted percutaneously.
Findings on clinical examination of the unoperated patient may include cyanosis, clubbing, a right ventricular lift, a thrill at the left sternal border due to severe pulmonary obstruction, or a loud continuous murmur over the thorax as a result of aortopulmonary collaterals in the setting of severe right ventricular outflow tract obstruction. After surgical repair, a diminished or an absent radial pulse (post Blalock–Taussig shunt), a soft ejection murmur across the right ventricular outflow tract, a low-pitched diastolic murmur from pulmonary regurgitation, or an absent P2 may be noted.
On the ECG of a patient with repaired TOF, a complete right bundle branch block is typically present. Chest radiography may be normal post-TOF repair; however, cardiomegaly may be seen in the setting of significant pulmonic and/or tricuspid valve regurgitation. A right aortic arch is often seen.
After repair of TOF, cardiomegaly as well as development of atrial or ventricular arrhythmia should prompt search for an underlying hemodynamic abnormality, commonly pulmonary regurgitation. Intervention on the pulmonary valve, whether surgically or percutaneously, is indicated in the setting of severe pulmonary regurgitation and associated symptoms, moderate-to-severe right ventricular dysfunction or enlargement, moderate-to-severe tricuspid regurgitation, or the development of symptomatic or sustained atrial or ventricular arrhythmias. Other indications for surgical intervention on patients with repaired TOF include residual right ventricular outflow tract obstruction, residual VSD (Qp/Qs > 1.5) and severe, symptomatic aortic regurgitation. As the RV may be suboptimally assessed by echocardiography, cardiac MRI can be useful to quantitatively evaluate the RV and aid in determining the ideal timing of intervention. It is also best at quantifying pulmonary regurgitation.
Ebstein Anomaly
Ebstein anomaly is the result of inferior displacement of the tricuspid valve into the RV, which results in “atrialization” of the RV As a result, the RV is very small and not infrequently hypocontractile. The posterior and septal leaflets of the tricuspid valve are often small and inadequate, while the anterior leaflet is very large and redundant, resembling a “sail.” The latter feature results in the characteristic “sail sound,” which occurs during closure of the tricuspid valve, followed by the tricuspid regurgitation murmur (if present). Over 50% of patients have either an ASD or a patent foramen ovale (PFO) present and right-to-left shunting through these defects results in cyanosis. About 25% of the patients have an accessory conduction pathway (Wolf–Parkinson–White syndrome). The age at presentation depends on the degree of anatomic and hemodynamic derangements. In adulthood, patients commonly present with exercise intolerance, dyspnea, fatigue, symptomatic arrhythmias, and right-sided heart failure.
The ECG of Ebstein patients shows very tall (Himalayan) P waves, which are a characteristic finding. Preexcitation may be noted as well as QRS prolongation with a “splintered” right bundle branch block pattern. Chest radiography may reveal marked cardiomegaly with clear lung fields. The cardiac contour tends to be “globular” due to right atrial enlargement. The diagnosis is typically confirmed using echocardiography. There is also an increasing role for cardiac MRI for clearer delineation of cardiac structure and function.
Surgery involves complex repair or replacement of the tricuspid valve in addition to closure of the atrial communication, and should be limited to centers with extensive experience in this area. Indications for surgery include significant symptoms or worsening exercise capacity, cyanosis (oxygen saturation <90%), progressive cardiomegaly on chest x-ray (often defined as a cardiothoracic ratio >60%), paradoxical embolism, and severe tricuspid regurgitation with progressive right ventricular dilation or dysfunction.
Eisenmenger Syndrome
As mentioned above, Eisenmenger physiology refers to the condition where an intracardiac shunt has resulted in extensive pulmonary vascular disease and pulmonary hypertension that is so severe that the shunt has reversed. The physical exam of these patients is notable for cyanosis (which often worsens during exercise) and clubbing. If differential clubbing is seen (usually clubbing of the feet and the left arm and not the right arm), then the clinical diagnosis is Eisenmenger physiology in the context of PDA. On physical exam, the jugular venous pressure is elevated; there may also be a right ventricular heave, a palpable P2 that is loud on auscultation, a systolic ejection click, and a diastolic murmur due to pulmonary regurgitation. Because pulmonary and systemic pressures only slightly differ, a systolic murmur across the lesion is generally not heard.
In general, patients with Eisenmenger syndrome have better long-term survival than comparable patients with idiopathic (primary) pulmonary hypertension, but their functional limitation is considerable. They may present with dyspnea on exertion, palpitations, progressive cyanosis, hemoptysis, syncope, or volume overload. Rapid deterioration can be seen during atrial or ventricular arrhythmias or with complications such pulmonary embolism or infection, or generally any condition that results in even transient hypotension.
There are a number of complications that result from long-standing hypoxia, including significant erythrocytosis (elevated red blood cell count). Symptoms of hyperviscosity (changes in mental status, fatigue, and headache) are quite rare, and phlebotomy should only be performsed to relieve these symptoms (typically in the presence of a hematocrit >65%) in the absence of dehydration. If phlebotomy is attempted, it should be accompanied by at least equal fluid replacement. Repeated phlebotomy can result in iron deficiency and actually increases the risk of hyperviscosity. Iron should be repleted in these patients if deficiency is present. Patients with Eisenmenger syndrome often develop proteinuria and a decreased glomerular filtration rate (GFR). Because of the low GFR and the high turnover of red blood cells, elevated uric acid levels are frequently seen and can result in acute renal failure, particular after administration of contrast dye if the patient is not adequately hydrated. Patients with Eisenmenger physiology are at risk of both thrombosis and hemorrhage, hemoptysis that may be life threatening, cerebral abscesses, stroke, scoliosis, and arthropathy as well as pigment gallstones. They may present with cardiac ischemia in the setting of coronary artery compression by a dilated PA, right ventricular ischemia, or atherosclerosis.
Patients with Eisenmenger physiology should avoid dehydration, moderate-to-severe strenuous exercise, exposure to excessive heat, chronic high altitude, and pregnancy. If catheterization or noncardiac surgery is required, they should be hospitalized in centers with adult CHD expertise and experienced cardiac anesthesia. All intravenous lines should be filtered to exclude air bubbles. Improved quality of life has been noted with the use of pulmonary vasodilators in patients with Eisenmenger physiology and survival may be positively impacted. Transplantation has also offered limited survival benefits albeit with significant quality of life improvement for this patient population.
GENERAL MANAGEMENT STRATEGIES
Although adult patients with CHD can be intimidating at first presentation, sticking to basic concepts can be helpful in choosing appropriate management strategies.
Patients with intracardiac shunts should be counseled to avoid high-risk activities such as scuba diving and have filtering devices placed on all intravenous lines whenever hospitalized to prevent the risk of paradoxical embolization. Noncardiac surgery should be considered on a patient- per-patient basis only after the risks and benefits have been carefully considered, particularly in the patient with Eisenmenger syndrome. Most adults with CHD will require lifelong follow-up and should be referred to tertiary care centers with expertise in their care.
Some patients with CHD are at increased risk of developing IE and should be educated on the recommendations for prophylaxis that were revised in 2007. According to the new guidelines, antibiotic prophylaxis before dental procedures is recommended to the “high-risk” group of patients with CHD including (1) those with prior IE; (2) those with prosthetic heart valves; (3) those with palliated or unrepaired cyanotic CHD, including surgically constructed palliative conduits and shunts; (4) those with repaired CHD with prosthetic material or device, whether placed percutaneously or surgically, during the first 6 months postprocedure; and (5) those with repaired CHD with residual defects at the site or adjacent to the site of a prosthetic device or patch that prevents endothelialization.
Approximately 18% of congenital heart defects are associated with a congenital syndrome, including coexisting cognitive and neurologic deficits or chromosomal abnormalities (Down syndrome with trisomy 21 and TOF with 22q11.2 deletion). These patients should be appropriately screened for coexisting noncardiac conditions affecting them including sleep apnea, endocrinopathies, renal disease, and psychiatric issues with appropriate referrals provided. Patients should also be counseled on important topics such as pregnancy, genetic counseling, and contraception. For illustrative cases of CHD lesions, please refer to Chapter 24.
Bashore TM. Adult congenital heart disease: right ventricular outflow tract lesions. Circulation. 2007;115:1933–1947.
Diller GP. Gatzoulis MA. Pulmonary vascular disease in adults with congenital heart disease. Circulation. 2007;115:1039–1050.
Rhodes JF, Hijazi ZM, Sommer RJ. Pathophysiology of congenital heart disease in the adult, part II. Simple obstructive lesions. Circulation. 2008;117:1228–1237.
Sommer RJ, Hijazi ZM, Rhodes JF Jr. Pathophysiology of congenital heart disease in the adult: part I: Shunt lesions. Circulation. 2008;117:1090–1099.
Sommer RJ, Hijazi ZM, Rhodes JF. Pathophysiology of congenital heart disease in the adult: part III: Complex congenital heart disease. Circulation. 2008;117:1340–1350.
Spence MS, Balaratnam MS, Gatzoulis MA. Clinical update: cyanotic adult congenital heart disease. Lancet. 2007;370:1530–1532.
Warnes CA. Adult congenital heart disease importance of the right ventricle. J Am Coll Cardiol. 2009;54:1903–1910.
Warnes CA, Williams RG, Bashore TM, et al. ACC/AHA 2008 Guidelines for the Management of Adults with Congenital Heart Disease: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (writing committee to develop guidelines on the management of adults with congenital heart disease). Circulation. 2008;118:2395–2451.
QUESTIONS AND ANSWERS
Questions
1. You are seeing a 27-year-old female postal employee in outpatient clinic. She has a history of asthma treated with inhalers, though her pulmonary function tests and methacholine challenge were recently normal. She has noted progressive fatigue over the past months and finds that getting up large hills on her mail route gets her out of breath. On examination, she has a fixed split second heart sound and soft systolic ejection murmur over the left upper sternal border. Her lungs are clear and all her extremity pulses are equal and of normal intensity. All the following would be expected to be present on her diagnostic studies except:
a. Unexplained right heart enlargement on echocardiography
b. An RSR’ (incomplete bundle branch block) pattern on electrocardiogram (ECG)
c. Unexplained mild pulmonary hypertension
d. Right-to-left shunt by bubble study on echocardiography
e. Decreased pulmonary vascularity on chest x-ray
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


