TABLE 140.1 Approximate Frequencies of Congenital Heart Diseases, as Proportion of Total | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||
TABLE 140.2 Syndromes and Congenital Heart Diseasea | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
TABLE 140.3 Recurrence Risk Siblings, Percent, After Diagnosis of Infant or Fetus with Congenital Heart Disease, by Type | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||
(both sides have typical right-sided features), and left isomerism (both sides have typical left-sided features). The spleen, being left sided, is absent in right-sided isomerism, on the right in situs inversus, and multiple in left-sided isomerism.
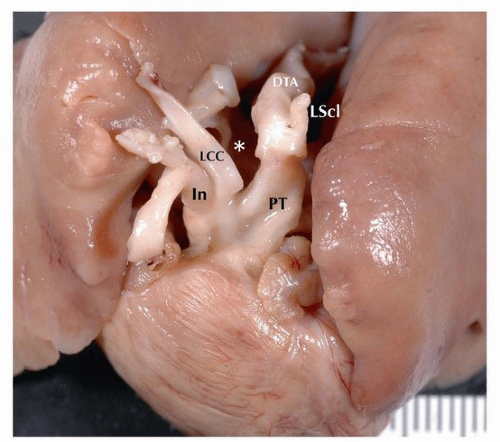 FIGURE 140.1 ▲ Interrupted aortic arch type B. LCC, left common carotid; PT, pulmonary trunk; LScl, left subclavian; DTA, descending thoracic aorta. Asterisk is the location of normal arch, which is absent. Interruption between left subclavian and left common carotid is in the B aortic segment, between the left common carotid and left subclavian. |
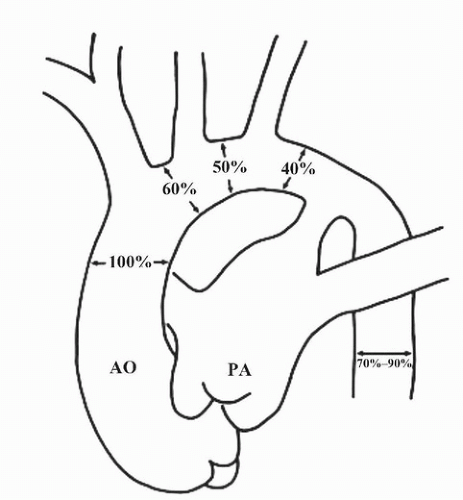 FIGURE 140.2 ▲ Normal diameters of the aortic arch in utero. Note that the isthmus is normally 40% that of the ascending aorta; narrowing of the isthmus should not be overcalled as coarctation. Reproduce with permission, Siewers RD, Ettedgui J, Pahl E, et al. Coarctation and hypoplasia of the aortic arch: will the arch grow? Ann Thorac Surg. 1991;52:608-614. |
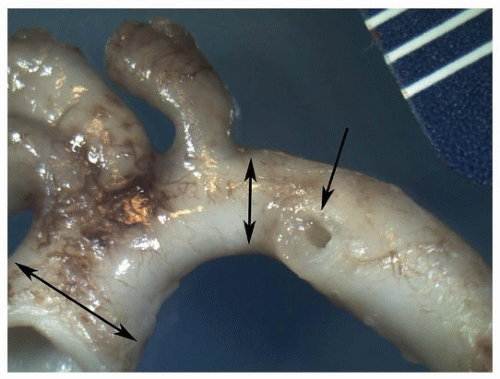 FIGURE 140.3 ▲ Normal infant aortic arch. Double arrows show ascending aorta (left) and isthmus (right). Single arrow shows the orifice of the ductus arteriosus. |
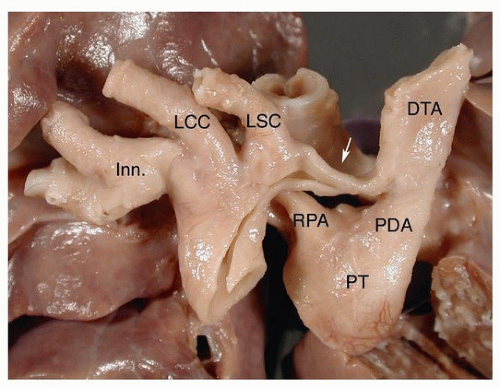 FIGURE 140.4 ▲ Aortic coarctation, preductal. The arrow points to the coarctation at the isthmus. Inn, innominate; LCC, left common carotid; LSC, left subclavian; RPA, right pulmonary artery; PT, pulmonary trunk; PDA, patent ductus arteriosus; DTA, descending thoracic aorta. |
tetralogy, persistent truncus arteriosus, and pulmonary atresia and ventricular septal defect have right-sided arches with mirror-image branching (see below), in addition to lower rates for double outlet right ventricle, tricuspid atresia, and transposition.
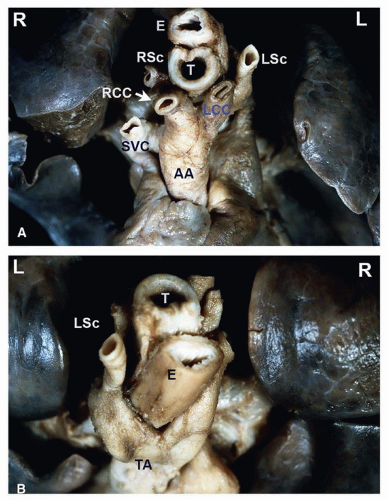 FIGURE 140.5 ▲ Double aortic arch, anterior/cranial view. A. The ascending aorta bifurcates into an anterior left branch, supplying the left common carotid artery and the left subclavian artery, and a posterior right branch, supplying the right common carotid and right subclavian arteries. B. Double aortic arch, posterior/cranial view. The continuation of the aorta viewed from behind demonstrates the anterior left branch wrapping around the trachea and esophagus as well as the right posterior branch emerging from under the esophagus. The distal aorta continues as a centrally located structure. Abbreviations: RCC, right common carotid artery; LCC, left common carotid artery; RSc, right subclavian artery; LSc, left subclavian artery; E, esophagus; T, trachea; AA, ascending aorta; TA, descending aorta; SVC, superior vena cava. |
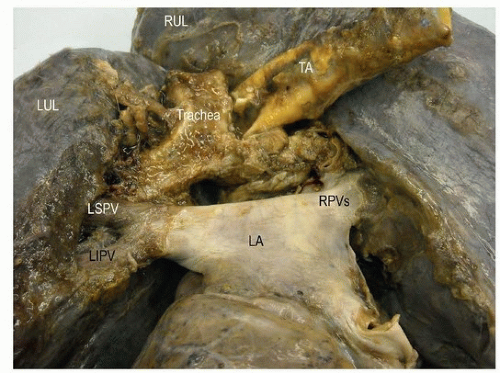 FIGURE 140.6 ▲ Right aortic arch, posterior view. The descending thoracic aorta (TA) is to the right of the trachea. RUL, right upper lobe; LUL, left upper lobe; LSPV, left superior pulmonary vein; RPV, right pulmonary vein; LA, left atrium. |
that may range from sporadic migraine to transient ischemic attacks or stroke. Transthoracic echocardiography with contrast or transesophageal echocardiography is usually required to detect small PFO.
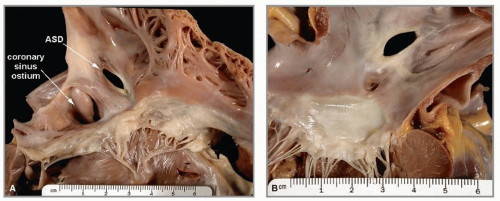 FIGURE 140.7 ▲ A. Atrial septal defect, secundum type, from right atrium. The atrial septal defect (ASD) is in the area of the oval fossa. The coronary sinus ostium appears dilated. B. Atrial septal defect, secundum type, from left atrium. This defect is the same as that illustrated in figure 8, from the left side. The anterior leaflet of the mitral valve is below. |
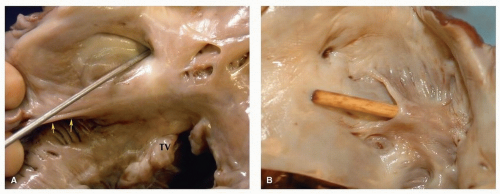 FIGURE 140.8 ▲ Patent foramen ovale, adult. A. The probe is present within the right side of the oval fossa. The eustachian valve is present at the bottom above the orifice of the coronary sinus. The septal leaflet of the tricuspid valve (TV) is present below. B. Viewed from the left, this patency was a tunnel within the foramen ovale. The characteristic “backward C” is present where the probe emerges. |
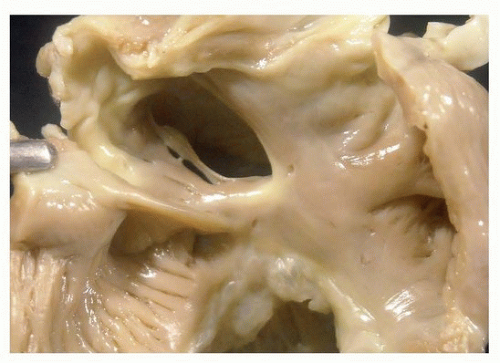 FIGURE 140.9 ▲ Atrial septal defect, secundum type, from the right. There is no septal tissue present in the oval fossa area, save a thin cord. The coronary sinus is present just below (posteriorly). |
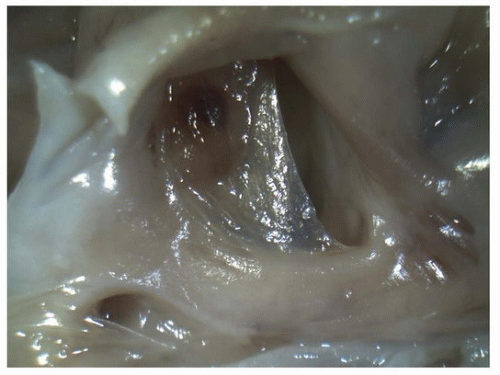 FIGURE 140.10 ▲ Patent foramen ovale, viewed from the right atrium. In this premature neonate, this is a normal finding. The oval fossa nearly covered by a thin, translucent membrane. It is easy to disrupt the ostium primum in fetal and neonatal hearts, and one should be careful not to overcall atrial septal defect. |
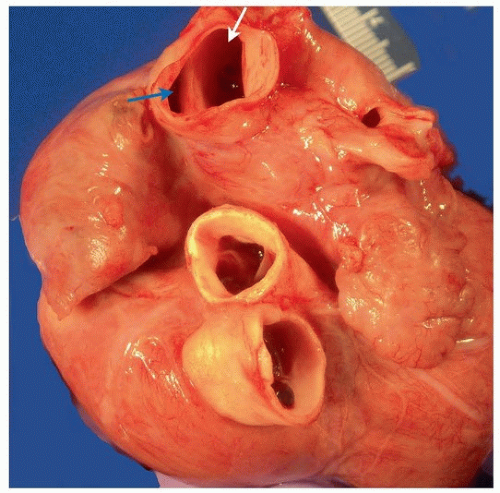 FIGURE 140.11 ▲ Sinus venosus atrial septal defect. The superior vena cava (above) is transected, revealing the superior rim of the atrial septal defect, exposing the right atrium (blue arrow) and left atrium (white arrow). |
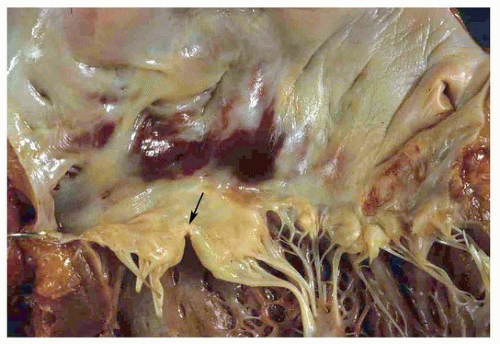 FIGURE 140.12 ▲ Partial atrioventricular canal defect. Note cleft of the anterior mitral valve leaflet (arrow). |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


