channels involved in ventricular repolarization such as those responsible for the fast component of the repolarizing transient outward K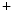 current (
current (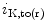 ) through the K
) through the K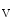 4.2–KChIP2 channel, thereby prolonging ventricular action potential duration (APD).2
4.2–KChIP2 channel, thereby prolonging ventricular action potential duration (APD).2
Artificial pacemakers are implanted to treat severe block syndromes. The pacemaker continuously monitors the heartbeat and delivers an imperceptible electrical charge to stimulate the heartbeat if needed. Typically, a pacemaker is endowed with two insulated wires (electrodes or leads) in the right atrium and ventricle (RA and RV leads). Cardiac resynchronization therapy (CRT) in congestive heart failure with electrical and mechanical dyssynchrony, especially in moderately to severely symptomatic patients despite medical therapy (dyspnea, edema of lower limb extremities, exercise intolerance, and depression; left ventricular ejection fraction [LVEF]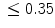 ), is aimed at restoring the normal coordinated pumping action of the ventricles by overcoming the delay in electrical conduction caused by a left bundle branch block (LBBB; prolonged QRS complex duration [
), is aimed at restoring the normal coordinated pumping action of the ventricles by overcoming the delay in electrical conduction caused by a left bundle branch block (LBBB; prolonged QRS complex duration [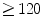 ms], [368]).
ms], [368]).
), is aimed at restoring the normal coordinated pumping action of the ventricles by overcoming the delay in electrical conduction caused by a left bundle branch block (LBBB; prolonged QRS complex duration [ ms], [368]).5.1 Summary of Electrochemical and Mechanical Events
Cardiac electrochemical events and involved ion carriers that trigger myocardial contraction and relaxation cycles have been described in Vols. 5 (Chap. 5. Cardiomyocytes) and 6 (Chap. 3. Cardiovascular Physiology). This section summarizes and updates data.
In a broad sense, opsins are light-sensitive proteins that include G-protein-coupled receptors (e.g., Gq-coupled melanopsin) and ion carriers. Optogenetics uses genetically encoded light-sensitive proteins to actuate and/or sense fast biological processes. Optogenetics pacing enables remote spatiotemporal exploration as well as control of cardiac cell excitability, signal transmission, and gene expression by light-induced transcriptional effectors [370–372]. However, inserted nonnative opsins may exert yet unknown effects and implantation of modified cells may disturb electrical coupling between cell types, in addition to a different electrophysiological behavior.
5.1.1 Electromechanical Coupling
The current transmitted between cardiomyocytes during discharge and small molecular messengers cross gap junctions made up of voltage-sensitive connexins (mainly Cx40, Cx43, and Cx45) and organized in arrays in intercalated discs. Gap junctions couple cardiomyocytes axially and transversely. Gap junction conductivity depends also on intracellular Ca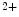 level, pH, and phosphorylation.
level, pH, and phosphorylation.
level, pH, and phosphorylation.The initial event in the cardiac cycle is the fast membrane depolarization, resulting from ion entry through gap junctions and opening of voltage-gated Na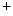 channels that induces Na
channels that induces Na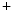 influx (Vol. 3, Chap. 3. Main Sets of Ion Channels and Pumps).
influx (Vol. 3, Chap. 3. Main Sets of Ion Channels and Pumps).
channels that induces Na influx (Vol. 3, Chap. 3. Main Sets of Ion Channels and Pumps).In myocytes, the Na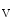 1.5 channel encoded by the SCN5A gene is the principal expressed voltage-gated Na
1.5 channel encoded by the SCN5A gene is the principal expressed voltage-gated Na channel. It is responsible for the largest membrane current in excitable cells that causes the rising phase of the action potential. Most voltage-gated Na
channel. It is responsible for the largest membrane current in excitable cells that causes the rising phase of the action potential. Most voltage-gated Na channels activate and inactivate completely within the first few milliseconds of an action potential. The Na
channels activate and inactivate completely within the first few milliseconds of an action potential. The Na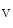 1.5 channel predominantly localizes to intercalated discs to successfully propagate action potentials.
1.5 channel predominantly localizes to intercalated discs to successfully propagate action potentials.
1.5 channel encoded by the SCN5A gene is the principal expressed voltage-gated Na channel. It is responsible for the largest membrane current in excitable cells that causes the rising phase of the action potential. Most voltage-gated Na channels activate and inactivate completely within the first few milliseconds of an action potential. The Na1.5 channel predominantly localizes to intercalated discs to successfully propagate action potentials.Membrane depolarization then inactivates Na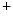 channels and opens both K
channels and opens both K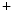 and Ca
and Ca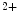 channels. Calcium entry into the cell triggers Ca
channels. Calcium entry into the cell triggers Ca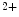 release from its stores (sarcoplasmic reticulum) via ryanodine-sensitive Ca
release from its stores (sarcoplasmic reticulum) via ryanodine-sensitive Ca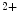 channels. Calcium ions then bind to troponin complexes and activate contractile sarcomeres.
channels. Calcium ions then bind to troponin complexes and activate contractile sarcomeres.
channels and opens both K and Ca channels. Calcium entry into the cell triggers Ca release from its stores (sarcoplasmic reticulum) via ryanodine-sensitive Ca channels. Calcium ions then bind to troponin complexes and activate contractile sarcomeres.Sarcomere relaxation follows upon Ca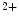 removal from the cytosol back into its stores by Ca
removal from the cytosol back into its stores by Ca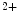 uptake pumps of the sarcoplasmic reticulum (SERCA) and extracellular milieu by sarcolemmal Na
uptake pumps of the sarcoplasmic reticulum (SERCA) and extracellular milieu by sarcolemmal Na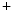 –Ca
–Ca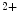 exchanger (NEX) and Ca
exchanger (NEX) and Ca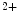 ATPases (PMCA). Intracellular Na
ATPases (PMCA). Intracellular Na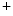 homeostasis is achieved by Na
homeostasis is achieved by Na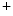 –K
–K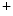 pumps.
pumps.
removal from the cytosol back into its stores by Ca uptake pumps of the sarcoplasmic reticulum (SERCA) and extracellular milieu by sarcolemmal Na–Ca exchanger (NEX) and Ca ATPases (PMCA). Intracellular Na homeostasis is achieved by Na–K pumps.5.1.2 Genesis and Propagation of Action Potentials—Nodal Tissue
Some cardiac cells are self-excitable, that is, have their own intrinsic gating and contraction rhythm. They pertains to the sinoatrial node, or natural pacemaker that sets the rate of contraction–relaxation cycles of cardiomyocytes (Vol. 5, Chap. 5. Cardiomyocytes). The sinoatrial node generates electrochemical impulses that are sent to neighboring electrically coupled cells via adhesion sites.
Action potentials spread rapidly through atrial walls and reach the atrioventricular node located between the right atrium and ventricle, where they are delayed for about 100 ms before traveling to the ventricular walls.
Action potentials then run through the His bundle and its branches and then Purkinje fibers that conduct signals from the cardiac apex to the basis. They produce electrical currents conducted to the skin, where these signals can be recorded as an electrocardiogram trace.
5.1.2.1 Sinoatrial Node
The sinusal or sinoatrial node (SAN; nodus sinuatrialis), also called Keith and Flack node, is an extensive structure within the right atrium that acts as the natural pacemaker (impulse generator) that emits action potentials with a normal sinusal rhythm.
Cardiac automaticity arises from the integrated activity of voltage-gated ionic currents (HCN2 and HCN4 as well as Ca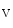 1.3 and Ca
1.3 and Ca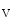 3.1), transporters (NCX), and sarcoplasmic reticulum Ca
3.1), transporters (NCX), and sarcoplasmic reticulum Ca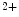 release.
release.
1.3 and Ca3.1), transporters (NCX), and sarcoplasmic reticulum Ca release.In addition to these classical ion carriers, some members of the TRP (transient receptor potential) superfamily of ion channels are also expressed in the myocardium (TRPC1, TRPC3–TRPC4, TRPC6, TRPM4, and TRPM7).
Structure and Location of the Sinoatrial Node
The sinoatrial node is cluster of specialized cardiomyocytes positioned on the inner layer of the right atrial wall near the entrance of the superior vena cava. It is a diffuse, but integrated aggregate of pacemaker cells that extends from the junction between the right atrium and superior vena cava (SVCRAJ) virtually to the junction between the right atrium and inferior vena cava (IVCRAJ) [373]. The right atrial pacemaker region comprises a zone of 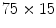 mm centered about the long axis of the sulcus terminalis (or crista terminalis) posteriorly and the precaval band anteriorly.
mm centered about the long axis of the sulcus terminalis (or crista terminalis) posteriorly and the precaval band anteriorly.
mm centered about the long axis of the sulcus terminalis (or crista terminalis) posteriorly and the precaval band anteriorly.The pacemaker is defined as unicentric when a single source of activation spreads to activate the atria. The pacemaker is considered as multicentric when several sites (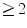 ) of early activation are separated by a distance greater than or equal to 10 mm with activation time difference shorter than or equal to 5 ms [374].
) of early activation are separated by a distance greater than or equal to 10 mm with activation time difference shorter than or equal to 5 ms [374].
) of early activation are separated by a distance greater than or equal to 10 mm with activation time difference shorter than or equal to 5 ms [374].Ca and HCN Channels
and HCN Channels
The spontaneous activity of sinoatrial myocytes determines cardiac frequency. The maximal cardiac frequency declines with aging, progressively limiting the aerobic capacity of the elderly, as the maximal cardiac frequency is a major determinant of age-dependent decrease in the maximum rate of oxygen consumption. The depressed spontaneous activity of sinoatrial myocytes results from altered ion channel activity [375]. These cells are characterized by:
These electophysiological changes are associated with cellular hypertrophy. The action potential upstroke, duration, and repolarization as well as the chronotropic response to β-adrenoceptor stimulation are preserved [375].
Slower firing rates
Altered action potential waveform with changes limited to hyperpolarization of the maximum diastolic potential and slowing of the early part of the diastolic depolarization

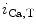
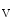

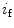
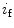
Na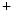 –Ca
–Ca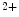 Exchanger
Exchanger
The spontaneous cardiac pacemaking relies on complex molecular mechanisms that include a coordinated interplay between intracellular Ca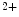 cycling and plasmalemmal cation transport. Among involved ion carriers, the Na
cycling and plasmalemmal cation transport. Among involved ion carriers, the Na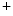 –Ca
–Ca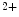 exchanger NCX1 is a sarcolemmal protein involved in the maintenance of calcium homeostasis in cardiomyocytes. Selective ablation of NCX1 in nodal cells provokes a progressive reduction of cardiac frequency with irregular firing accompanied by severe bradycardia as well as various supraventricular and ventricular rhythm disorders (sinusal arrhythmia and pauses, atrioventricular block, and ventricular tachycardia, [376]). In addition, Ca
exchanger NCX1 is a sarcolemmal protein involved in the maintenance of calcium homeostasis in cardiomyocytes. Selective ablation of NCX1 in nodal cells provokes a progressive reduction of cardiac frequency with irregular firing accompanied by severe bradycardia as well as various supraventricular and ventricular rhythm disorders (sinusal arrhythmia and pauses, atrioventricular block, and ventricular tachycardia, [376]). In addition, Ca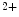 transients have a smaller magnitude and slower kinetics.
transients have a smaller magnitude and slower kinetics.
cycling and plasmalemmal cation transport. Among involved ion carriers, the Na–Ca exchanger NCX1 is a sarcolemmal protein involved in the maintenance of calcium homeostasis in cardiomyocytes. Selective ablation of NCX1 in nodal cells provokes a progressive reduction of cardiac frequency with irregular firing accompanied by severe bradycardia as well as various supraventricular and ventricular rhythm disorders (sinusal arrhythmia and pauses, atrioventricular block, and ventricular tachycardia, [376]). In addition, Ca transients have a smaller magnitude and slower kinetics.The NCX1 loss in pacemaker cells is not compensated by an modified expression of PMCA and Ca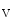 channels. The associated increase in the production of SERCA2 is not translated into an amplified Ca
channels. The associated increase in the production of SERCA2 is not translated into an amplified Ca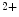 sequestration in the sarcoplasmic reticulum. Pacemaker cells have an elevated cAMP level and a pronounced basal PKA activity very likely due to the constitutive activity of calcium-activated adenylate cyclases AC1 and AC8 [376]. Pacemaker cells lacking NCX1 have reduced, slower, and disrhythmic Ca
sequestration in the sarcoplasmic reticulum. Pacemaker cells have an elevated cAMP level and a pronounced basal PKA activity very likely due to the constitutive activity of calcium-activated adenylate cyclases AC1 and AC8 [376]. Pacemaker cells lacking NCX1 have reduced, slower, and disrhythmic Ca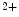 oscillations. In addition, the responsiveness to ligands of Gi-coupled receptors increases, but that of agonists targeting Gs-coupled receptors decreases. Hence, β-adrenergic response amplitude decays, whereas the adenosinergic and muscarinic effect intensity augment [376]. Gi- and Gs-coupled receptors inactivate and activates PKA, respectively. PKA phosphorylates phospholamban, an inhibitor of SERCA2 density, hence relieving this inhibition. The combined effect of attenuated AC activity and PKA inactivation may counterbalance increased SERCA2 level in NCX1-lacking pacemaker cells.
oscillations. In addition, the responsiveness to ligands of Gi-coupled receptors increases, but that of agonists targeting Gs-coupled receptors decreases. Hence, β-adrenergic response amplitude decays, whereas the adenosinergic and muscarinic effect intensity augment [376]. Gi- and Gs-coupled receptors inactivate and activates PKA, respectively. PKA phosphorylates phospholamban, an inhibitor of SERCA2 density, hence relieving this inhibition. The combined effect of attenuated AC activity and PKA inactivation may counterbalance increased SERCA2 level in NCX1-lacking pacemaker cells.
channels. The associated increase in the production of SERCA2 is not translated into an amplified Ca sequestration in the sarcoplasmic reticulum. Pacemaker cells have an elevated cAMP level and a pronounced basal PKA activity very likely due to the constitutive activity of calcium-activated adenylate cyclases AC1 and AC8 [376]. Pacemaker cells lacking NCX1 have reduced, slower, and disrhythmic Ca oscillations. In addition, the responsiveness to ligands of Gi-coupled receptors increases, but that of agonists targeting Gs-coupled receptors decreases. Hence, β-adrenergic response amplitude decays, whereas the adenosinergic and muscarinic effect intensity augment [376]. Gi- and Gs-coupled receptors inactivate and activates PKA, respectively. PKA phosphorylates phospholamban, an inhibitor of SERCA2 density, hence relieving this inhibition. The combined effect of attenuated AC activity and PKA inactivation may counterbalance increased SERCA2 level in NCX1-lacking pacemaker cells.Transient Receptor Potential Melastatin (TRPM) Channel
TRPM7, a divalent-permeant channel-kinase, is highly expressed in the sinoatrial node, where it is required for cardiac automaticity, in addition to human atrial myocytes and fibroblasts [377]. It enables outwardly rectifying current that is inhibited by both cytoplasmic and extracellular Mg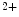 ions.
ions.
ions. In atrial fibrillation, the TRPM7-mediated current is elevated in human atriofibroblasts [377]. Ventricular fibroblasts are also sites of large TRPM7-conveyed currents. The TRPM7 channel concentration is the largest in myocardial cells endowed with automaticity.
Ablation of the TRPM7 channel in cultured embryonic cardiomyocytes significantly reduces spontaneous Ca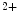 transient firing rates as well as production of Hcn4, CACNA1G (Ca
transient firing rates as well as production of Hcn4, CACNA1G (Ca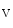 3.1), and SERCA2A mRNAs. Global murine cardiac TRPM7 deletion disrupts cardiac automaticity in vivo, causing sinusal pauses and atrioventricular node block. The TRPM7 channel influences diastolic membrane depolarization and automaticity in SAN cells via the Hcn4 gene transcription [377]. It may operate via the transcription factors Tbx3 and/or MEF2.
3.1), and SERCA2A mRNAs. Global murine cardiac TRPM7 deletion disrupts cardiac automaticity in vivo, causing sinusal pauses and atrioventricular node block. The TRPM7 channel influences diastolic membrane depolarization and automaticity in SAN cells via the Hcn4 gene transcription [377]. It may operate via the transcription factors Tbx3 and/or MEF2.
transient firing rates as well as production of Hcn4, CACNA1G (Ca3.1), and SERCA2A mRNAs. Global murine cardiac TRPM7 deletion disrupts cardiac automaticity in vivo, causing sinusal pauses and atrioventricular node block. The TRPM7 channel influences diastolic membrane depolarization and automaticity in SAN cells via the Hcn4 gene transcription [377]. It may operate via the transcription factors Tbx3 and/or MEF2.Another TRPM family member, TRPM4, a Ca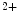 -activated, Na
-activated, Na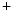 -selective channal is enriched in human Purkinje cells. Mutations (gain-of-function) of the Trpm4 gene is responsible for the progressive familial heart block-I associated with an impaired TRPM4 endocytosis secondary to constitutive sumoylation.
-selective channal is enriched in human Purkinje cells. Mutations (gain-of-function) of the Trpm4 gene is responsible for the progressive familial heart block-I associated with an impaired TRPM4 endocytosis secondary to constitutive sumoylation.
-activated, Na-selective channal is enriched in human Purkinje cells. Mutations (gain-of-function) of the Trpm4 gene is responsible for the progressive familial heart block-I associated with an impaired TRPM4 endocytosis secondary to constitutive sumoylation.5.1.2.2 Atrioventricular Node
The atrioventricular node conducts slowly and has a long refractory period, thereby engendering a delay between atrial and His bundle activation, controlling the ventricular impulse rate, and filtering atrial impulses especially during supraventricular tachyarrhythmias. The atrioventricular node is characterized by rate-dependent conduction and refractoriness [378]. However, the atrioventricular node with its fast and slow pathway is also the site of atrioventricular nodal reentrant tachycardia, the most common form of paroxysmal supraventricular tachycardia.
The atrioventricular node occupies the Koch triangle, the upper atrial and lower ventricular sides of which correspond to the tendon of Todaro and tricuspid valve insertion, respectively. The His bundle and coronary sinus ostium represent its anterosuperior apex and posteroinferior base, respectively.
The atrioventricular node contains substructures (transitional zone, posterior extension, compact node, and lower nodal bundle) that determine conduction time [378]. The connection between the transitional zone, compact node, and lower nodal bundle is related to fast conduction pathway. Another connection between the septal portion of the transitional zone and lower nodal bundle bypassing the compact node may also yield another fast conduction pathway. The posterior transitional zone, posterior extension, and lower bundle together provide the slow conduction pathway. The transitional zone that connects the atrium to the compact node and posterior extension gives the common proximal pathway. The lower nodal bundle is linked to both the compact node and posterior extension and furnishes the common distal pathway to the His bundle.
The atrioventricular node function is assessed with various protocols, each with their own drawbacks, such as the premature or extrastimulus protocol to identify the recovery and fatigue properties [378].
5.1.3 Myocardial Mechanics and Electrical Activity
Action potentials are electrochemical impulses that trigger myocardial contraction (excitation–contraction coupling [ECC]). Conversely, mechanical state of the myocardium influences its electrical activity (mechanoelectrical [MEC] or contraction–excitation [CEC] coupling, [379]).
The shape of action potentials can be modified by four basic mechanisms [379]:
A diastolic elevated stretch can provoke an immediate reversible depolarization. The strain field may affect the ion carrier kinetics and change their permeability. When stretched myofibers are stimulated, the action potential from partly depolarized membrane has a reduced amplitude. In addition, the conduction velocity lowers at least in stretched rat ventricles.
1.
The degree of inactivation or recovery of ionic carriers
2.
The concentrations of cytosolic Ca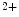 and external K
and external K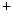 ions
ions
and external K ions3.
The level of ionic exchanges, in particular through Na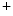 –K
–K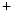 and Na
and Na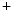 –Ca
–Ca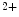 exchangers
exchangers
–K and Na–Ca exchangers4.
The stretch magnitude, which can modify the resting transmembrane potential
5.1.4 Sarcolemmal Density of Ion Carriers
Ion channels, exchangers, and pumps yield inward excitatory and/or outward repolarizing currents. Normal cardiac electrochemical activity depends on the expression with a proper density of numerous ion carriers and their partners in specialized nanodomains of the sarcolemma and organelles of cardiomyocytes.
The plasmalemmal density of ion carriers depends on the transfer of their subunits from the endoplasmic reticulum and Golgi body to the cell surface. Exo- and endocytosis that continuously regulate the degradation or recycling of ion carriers control the density of functional ion channels in response to various stimuli.
5.1.4.1 K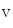 Channels
Channels
The density of K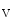 11.1 channels is also regulated by external potassium concentrations [380]. In LQTS2 syndrome due to a missense mutation in KCNH2 gene encoding K
11.1 channels is also regulated by external potassium concentrations [380]. In LQTS2 syndrome due to a missense mutation in KCNH2 gene encoding K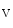 11.1 channel (responsible for the rapid delayed rectifier K
11.1 channel (responsible for the rapid delayed rectifier K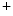 current [
current [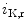 ]) associated with a prolonged ventricular repolarization, the defective channel transfer to the sarcolemma results from an elevated transport of the mutated, but functional channel to the proteasome [381].
]) associated with a prolonged ventricular repolarization, the defective channel transfer to the sarcolemma results from an elevated transport of the mutated, but functional channel to the proteasome [381].
11.1 channels is also regulated by external potassium concentrations [380]. In LQTS2 syndrome due to a missense mutation in KCNH2 gene encoding K11.1 channel (responsible for the rapid delayed rectifier K current []) associated with a prolonged ventricular repolarization, the defective channel transfer to the sarcolemma results from an elevated transport of the mutated, but functional channel to the proteasome [381].In atriomyocytes, the surface expression of the main repolarizing channel K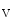 1.5 depends on the working conditions of the atrium. Hemodynamic stress stimulates the delivery of K
1.5 depends on the working conditions of the atrium. Hemodynamic stress stimulates the delivery of K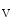 1.5 channels from recycling endosomes via an Itg–FAK-mediated exocytosis [380]. During atrial pressure overload, this process is upregulated; the plasmalemmal K
1.5 channels from recycling endosomes via an Itg–FAK-mediated exocytosis [380]. During atrial pressure overload, this process is upregulated; the plasmalemmal K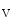 1.5 density rises and contributes to the shortening of the APD.
1.5 density rises and contributes to the shortening of the APD.
1.5 depends on the working conditions of the atrium. Hemodynamic stress stimulates the delivery of K1.5 channels from recycling endosomes via an Itg–FAK-mediated exocytosis [380]. During atrial pressure overload, this process is upregulated; the plasmalemmal K1.5 density rises and contributes to the shortening of the APD.5.1.4.2 SK Channels
Small-conductance Ca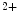 -activated K
-activated K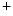 channels (SK; K
channels (SK; K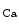 2) are widespread throughout the body. In excitable cells, they aid in processing changes in cytosolic Ca
2) are widespread throughout the body. In excitable cells, they aid in processing changes in cytosolic Ca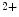 concentration and ensuring proper membrane potentials. The SK channels are gated directly by submicromolar concentrations of calcium. In humans, cardiomyocytes express all three SK channel subtypes (SK1–SK3 or K
concentration and ensuring proper membrane potentials. The SK channels are gated directly by submicromolar concentrations of calcium. In humans, cardiomyocytes express all three SK channel subtypes (SK1–SK3 or K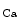 2.1–K
2.1–K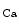 2.3). They can heteromultimerize. The SK2 subtype is highly sensitive to apamin, whereas both SK1 and SK3 channels show intermediate sensitivity. In atria, the SK channel contributes to repolarization.
2.3). They can heteromultimerize. The SK2 subtype is highly sensitive to apamin, whereas both SK1 and SK3 channels show intermediate sensitivity. In atria, the SK channel contributes to repolarization.
-activated K channels (SK; K2) are widespread throughout the body. In excitable cells, they aid in processing changes in cytosolic Ca concentration and ensuring proper membrane potentials. The SK channels are gated directly by submicromolar concentrations of calcium. In humans, cardiomyocytes express all three SK channel subtypes (SK1–SK3 or K2.1–K2.3). They can heteromultimerize. The SK2 subtype is highly sensitive to apamin, whereas both SK1 and SK3 channels show intermediate sensitivity. In atria, the SK channel contributes to repolarization.The SK density is larger in atriomyocytes than in ventriculomyocytes [382]. The SK1 and SK2 channel subtypes are predominantly expressed in the atrium and SK3 is expressed in the atrium and the ventricle [383]. Levels of SK2 and SK3 transcripts are higher than that of the SK1 subtype in human atria [384].
The members of the SK channel family are encoded by the genes of the human KCNN family (KCNN1–KCNN4), the SK4 channel being in fact the intermediate-conductance IK channel (IK1 [IK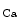 1] or K
1] or K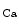 3.1 protein). The IK
3.1 protein). The IK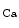 1 channel is an important contributor of the repolarization maintenance, at least in a rabbit heart failure model [383].
1 channel is an important contributor of the repolarization maintenance, at least in a rabbit heart failure model [383].
1] or K3.1 protein). The IK1 channel is an important contributor of the repolarization maintenance, at least in a rabbit heart failure model [383].The SK channel accelerates atrial repolarization, thereby augmenting the risk of atrial fibrillation from the pulmonary vein outlet region [385].
Both SK2 and SK3 levels are reduced in patients with chronic atrial fibrillation (CAF) with respect to patients with sinusal rhythm (SR). In isolated atriomyocytes from SR patients, a SK-selective inhibitor diminishes the inwardly rectifying K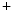 current by about 15 % and prolonges APD, without affecting myocytes from CAF patients [384]. In trabeculae muscular strips from right atrial appendages from SR patients, a SK-selective inhibitor raises APD and effective refractory period and depolarizes the resting membrane potential, but only a SK-slightly selective inhibitor provokes these effects in CAF patients [384]. On the other hand, the SK channels do not contribute substantially to the ventricular action potential.
current by about 15 % and prolonges APD, without affecting myocytes from CAF patients [384]. In trabeculae muscular strips from right atrial appendages from SR patients, a SK-selective inhibitor raises APD and effective refractory period and depolarizes the resting membrane potential, but only a SK-slightly selective inhibitor provokes these effects in CAF patients [384]. On the other hand, the SK channels do not contribute substantially to the ventricular action potential.
current by about 15 % and prolonges APD, without affecting myocytes from CAF patients [384]. In trabeculae muscular strips from right atrial appendages from SR patients, a SK-selective inhibitor raises APD and effective refractory period and depolarizes the resting membrane potential, but only a SK-slightly selective inhibitor provokes these effects in CAF patients [384]. On the other hand, the SK channels do not contribute substantially to the ventricular action potential.In KCNN2 (SK2) knockout mice, atrial repolarization is disturbed with elevated susceptibility to atrial fibrillation [386]. Variants in the KCNN3 transcript gene encoding the SK3 subtype (K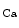 2.3) augments the risk of atrial fibrillation [386].
2.3) augments the risk of atrial fibrillation [386].
2.3) augments the risk of atrial fibrillation [386].The expression of SK channels in canine atria depends on the atrial region and wall remodeling. SK channel block does not cause statistically significant effects on APD in the left atrium, but affects it in pulmonary vein cardiomyocyte sleeves, especially in the presence of atrial fibrillation-related remodeling [386].
In homozygote SK3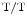 mice, overexpression of the SK3 channel abbreviates atrial APD and facilitates occurrence of atrial fibrillation. The APD is significantly shortened at 90 % repolarization (APD
mice, overexpression of the SK3 channel abbreviates atrial APD and facilitates occurrence of atrial fibrillation. The APD is significantly shortened at 90 % repolarization (APD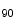 ) in atriomyocytes, but not at 50 % (conserved APD
) in atriomyocytes, but not at 50 % (conserved APD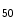 , [382]). In addition, overexpressed SK3 channels does not alter the resting membrane potentials.
, [382]). In addition, overexpressed SK3 channels does not alter the resting membrane potentials.
mice, overexpression of the SK3 channel abbreviates atrial APD and facilitates occurrence of atrial fibrillation. The APD is significantly shortened at 90 % repolarization (APD) in atriomyocytes, but not at 50 % (conserved APD, [382]). In addition, overexpressed SK3 channels does not alter the resting membrane potentials. In homozygote (SK3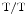 ) and heterozygote (SK3
) and heterozygote (SK3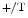 ) SK3-overexpressing mice, SK3 overexpression also disturbs conduction at and below the atrioventricular node [383]. In SK3
) SK3-overexpressing mice, SK3 overexpression also disturbs conduction at and below the atrioventricular node [383]. In SK3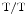 mice, cardiac conduction blocks and bradyarrhythmias may cause premature sudden death with abnormal atrioventricular node morphology (neurological or respiratory dysfunction can be actual cause of death). Ventricular conduction slows down by 25 % with respect to wild-type (WT) controls (
mice, cardiac conduction blocks and bradyarrhythmias may cause premature sudden death with abnormal atrioventricular node morphology (neurological or respiratory dysfunction can be actual cause of death). Ventricular conduction slows down by 25 % with respect to wild-type (WT) controls (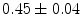 vs.
vs. 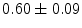 mm/ms in WT mice). At 1 month age, the atrioventricular nodal refractory period lowers (
mm/ms in WT mice). At 1 month age, the atrioventricular nodal refractory period lowers (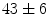 vs.
vs. 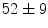 ms [decrease of 18 %]); at 3 months, the atrioventricular nodal refractoriness is prolonged (
ms [decrease of 18 %]); at 3 months, the atrioventricular nodal refractoriness is prolonged (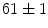 vs.
vs. 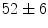 ms [increase of 17 %]); at 5 months, the atrioventricular node enlarges and is disorganized [383].
ms [increase of 17 %]); at 5 months, the atrioventricular node enlarges and is disorganized [383].
) and heterozygote (SK3) SK3-overexpressing mice, SK3 overexpression also disturbs conduction at and below the atrioventricular node [383]. In SK3 mice, cardiac conduction blocks and bradyarrhythmias may cause premature sudden death with abnormal atrioventricular node morphology (neurological or respiratory dysfunction can be actual cause of death). Ventricular conduction slows down by 25 % with respect to wild-type (WT) controls ( vs. mm/ms in WT mice). At 1 month age, the atrioventricular nodal refractory period lowers ( vs. ms [decrease of 18 %]); at 3 months, the atrioventricular nodal refractoriness is prolonged ( vs. ms [increase of 17 %]); at 5 months, the atrioventricular node enlarges and is disorganized [383].5.1.5 Complexes Incorporating Ion Channels
The spatial dynamical organization of ion channels and adaptor proteins in molecular complexes in cardiomyocytes can confer specific roles to these complexes in the generation and/or propagation of action potentials.
Numerous PKA-sensitive ion channel complexes contain A-kinase-anchoring protein that recruits kinases, phosphatases, and phosphodiesterases to control the local phosphorylation state.
Voltage-gated sodium channels complex with the ankyrin, calmodulin, dystrophin, syntrophin, fibroblast growth factor homologous factor FHF1b,6 and NEDD4-like protein–ubiquitin ligase. These partners are involved in the regulation of channel synthesis, localization, activity, and degradation of the Na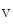 α subunit [388].
α subunit [388].
α subunit [388]. Calcium–calmodulin kinase-2 (CamK2α–CamK2δ) regulates ion channel functioning, especially voltage-gated Na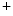 channels primarily in cardiomyocytes. It also phosphorylates Ca
channels primarily in cardiomyocytes. It also phosphorylates Ca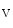 1.2a channel β2a subunit on the T-tubule in cardiomyocytes.
1.2a channel β2a subunit on the T-tubule in cardiomyocytes.
channels primarily in cardiomyocytes. It also phosphorylates Ca1.2a channel β2a subunit on the T-tubule in cardiomyocytes.In cardiomyocytes, the actin-binding protein β4-spectrin regulates the insertion of ion channels into the intercalated disc, as it connects to the adaptor protein ankyrin-G [389].
The anchoring protein β4-spectrin can have opposite effects on cardiac repolarization according to whether it interacts with K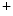 channel or Na
channel or Na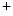 channel.
channel.
channel or Na channel.β4-Spectrin links to the 2-pore K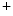 channel TREK1, which is environmental and physical factors (membrane stretch, pH, and polyunsaturated fatty acids). It is activated by mechanical stress and arachidonic acid accumulation. It then participates in action potential shaping. In mice, in the absence of β4-spectrin–TREK1 binding, TREK1 channel is aberrantly located and its activity decays, hence causing delayed action potential repolarization and arrhythmia [389].
channel TREK1, which is environmental and physical factors (membrane stretch, pH, and polyunsaturated fatty acids). It is activated by mechanical stress and arachidonic acid accumulation. It then participates in action potential shaping. In mice, in the absence of β4-spectrin–TREK1 binding, TREK1 channel is aberrantly located and its activity decays, hence causing delayed action potential repolarization and arrhythmia [389].
channel TREK1, which is environmental and physical factors (membrane stretch, pH, and polyunsaturated fatty acids). It is activated by mechanical stress and arachidonic acid accumulation. It then participates in action potential shaping. In mice, in the absence of β4-spectrin–TREK1 binding, TREK1 channel is aberrantly located and its activity decays, hence causing delayed action potential repolarization and arrhythmia [389].β4-Spectrin also tethers to the CamK2 kinase and the cardiac Na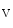 1.5 and contributes to the regulation of APD [390]. Inward Na
1.5 and contributes to the regulation of APD [390]. Inward Na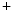 current quickly inactivate to allow repolarization. Ankyrin-G recruited by β4-spectrin brings Na
current quickly inactivate to allow repolarization. Ankyrin-G recruited by β4-spectrin brings Na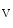 1.5 complexed with the actin–spectrin cytoskeleton at intercalated discs. The CamK2δ kinase phosphorylates (Ser571) the Na
1.5 complexed with the actin–spectrin cytoskeleton at intercalated discs. The CamK2δ kinase phosphorylates (Ser571) the Na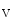 1.5 channel. Deletion of the CamK2-binding site of β4-spectrin eliminates the late Na
1.5 channel. Deletion of the CamK2-binding site of β4-spectrin eliminates the late Na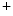 current and causes a marked shortening of the action potential.
current and causes a marked shortening of the action potential.
1.5 and contributes to the regulation of APD [390]. Inward Na current quickly inactivate to allow repolarization. Ankyrin-G recruited by β4-spectrin brings Na1.5 complexed with the actin–spectrin cytoskeleton at intercalated discs. The CamK2δ kinase phosphorylates (Ser571) the Na1.5 channel. Deletion of the CamK2-binding site of β4-spectrin eliminates the late Na current and causes a marked shortening of the action potential.Many other scaffolding or anchoring proteins interacts with various ion channels in cardiomyocytes [380].
The membrane-associated guanylate kinase (MAGuK) protein Disc large homolog DLg1 regulates the formation of the Na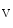 1.5–K
1.5–K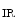 2.1 complex that may be involved in depolarization (rising phase of action potential). The K
2.1 complex that may be involved in depolarization (rising phase of action potential). The K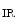 2.1 channel may maintain surrounding Na
2.1 channel may maintain surrounding Na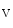 1.5 channels in the closed state.
1.5 channels in the closed state.
1.5–K2.1 complex that may be involved in depolarization (rising phase of action potential). The K2.1 channel may maintain surrounding Na1.5 channels in the closed state. Catenin-γ supports the formation of the Cx43–Na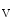 1.5 complex at the desmosome.
1.5 complex at the desmosome.
1.5 complex at the desmosome.The cardiac Na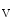 1.5 channel can also tether to the dystrophin-associated glycoprotein complex.
1.5 channel can also tether to the dystrophin-associated glycoprotein complex.
1.5 channel can also tether to the dystrophin-associated glycoprotein complex.5.1.6 Cardiomyocyte Tubules
The plasma membrane of cardiomyocytes invaginates to form the tubular set involved in excitation–contraction coupling. The tubular set is characterized by transverse (T) and longitudinal (or axial [A]) tubules running from a T tubule to the next. Ion carriers that respond to action potentials localize to the tubular set with different densities and isoforms from those in the remaining part of the cardiomyocyte sarcolemma. Action potentials are identical in amplitude and duration throughout the entire sarcolemma, i.e., in tubules and extratubular cell surface [391]. The myocyte tubular set rapidly conducts depolarization to trigger Ca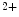 entry across the sarcolemma that initiates Ca
entry across the sarcolemma that initiates Ca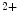 release from the sarcoplasmic reticulum, thereby priming synchronous contraction of the sarcomere set in the entire myocyte.
release from the sarcoplasmic reticulum, thereby priming synchronous contraction of the sarcomere set in the entire myocyte.
entry across the sarcolemma that initiates Ca release from the sarcoplasmic reticulum, thereby priming synchronous contraction of the sarcomere set in the entire myocyte.A structural disorganization of the tubular set alters the electrical coupling between it and the remaining part of the cardiomyocyte surface. Pathological remodeling of the tubular set can prime asynchronous calcium influx, hence dysfunctional cardiac contractility and arrhythmias [391].
5.2 Arrhythmias
Abnormal heart rhythms, notably atrial and ventricular fibrillation, cause death (10–20 % of all deaths among adults in the developed world). Arrhythmias result from interactions between a susceptible anatomical region (e.g., myocardial scar and fibrosis) and a triggering event (e.g., adrenergic surge, inflammation, acute myocardial ischemia, unusual wall stretch, and drug administration) [392]. Heart diseases with remodeling and altered expression and/or function of ion carriers frequently generate episodes of arrhythmias.
Magnetic resonance imaging with tagging can evaluate pacing protocols and locate ectopic sites . The temporal evolution of 3D strain fields has been computed from MR tagging of canine hearts for three different pacing sites, the base of the left ventricle free wall, the right ventricular apex, and the right atrium [393]. Right atrium pacing shows rapid synchronous shortening. Left ventricle base pacing exhibits a slowly propagating wavefront from the pacing site. Right ventricle apex pacing induces regional variations in propagation, with rapid septal activation followed by slower than usual activation. The propagation speed corresponds to the speed of electrochemical propagation in the myocardium.
During catheterization, using either a multielectrode basket (contact mapping system) or wire mesh, twinned with a second radiofrequency emitting catheter (noncontact mapping system), magnetic resonance, and mobile X-ray imaging can be merged into a common coordinate system using tracking and registration (XMR) to collect cardiac anatomy, motion, and electrical activity [394]. In vivo observations can then be combined with numerical simulations of the heart pathological rhythms.
5.2.1 Types of Arrhythmias
Heterogeneities in excitability and/or conduction velocities in the heart wall can initiate arrhythmias. Furthermore, increased dispersion of recovery and/or refractoriness (Vol. 5, Chaps. 5. Cardiomyocytes and 6. Heart Wall) in nodal cells caused by tissue damage can produce a conduction block. A premature impulse from an area with a short refractory period can propagate and find a region with a longer refractory period, leading to unidirectional propagation.
5.2.1.1 Bradycardia and Tachycardia
5.2.1.2 Long- or Short-QT Syndromes
5.2.1.3 Early and Delayed Afterdepolarizations
Alterations of the electric properties of cardiomyocytes in heart failure can favor the occurrence of atrial and ventricular arrhythmias by inducing EADs or DADs.
Arrhythmias can spontaneously arise in hearts that beat at normal or slow rates, i.e., not necessarily at fast cardiac frequency, although EADs can also appear at rapid cardiac frequencies, often in association with DADs.
In heart diseases, cardiomyocytes exhibit abnormal electrical oscillations such as EADs generated by drugs or due to genetic modifications. These coupled oscillators can synchronize and then propagate depolarizations. EADs at slow cardiac frequency actually exhibit chaotic dynamics [396].
Ventricular tachycardia or fibrillation results from spontaneous, premature ventricular complexes that typically occur irregularly (not randomly, but in the framework of dynamical chaos) and can be caused by EADs that self-organize. However, at least partial synchronization is required for propagation of EADs. Chaotic EADs globally synchronize when the tissue volume is smaller than a critical size [396].
On the other hand, when the tissue size exceeds the critical threshold, regions of partial synchronization generate premature ventricular complexes. These complexes propagate into recovered tissue without EADs.
Early Afterdepolarizations
EADs can result from a secondary activation of the Ca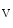 1.2 channel during the plateau of the action potential, as well as decreased activity from the rapid component of the delayed rectifier K
1.2 channel during the plateau of the action potential, as well as decreased activity from the rapid component of the delayed rectifier K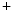 channels (K
channels (K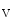 11.1) in damaged cardiomyocytes. EADs can result also from both
11.1) in damaged cardiomyocytes. EADs can result also from both 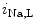 , in addition to
, in addition to 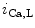 currents [397].
currents [397].
1.2 channel during the plateau of the action potential, as well as decreased activity from the rapid component of the delayed rectifier K channels (K11.1) in damaged cardiomyocytes. EADs can result also from both , in addition to currents [397].The late Na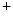 current (
current (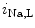 ) is indeed arrhythmogenic, as it induces an prolonged inward current favorable for EADs. Intracellular Na
) is indeed arrhythmogenic, as it induces an prolonged inward current favorable for EADs. Intracellular Na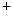 cycling is coupled with Ca
cycling is coupled with Ca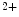 homeostasis, as Na
homeostasis, as Na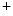 modulates the transport direction of the Na
modulates the transport direction of the Na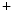 –Ca
–Ca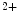 exchanger. Cellular Na
exchanger. Cellular Na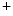 overload as well as action potential prolongation stimulates the reverse mode of Na
overload as well as action potential prolongation stimulates the reverse mode of Na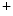 –Ca
–Ca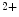 exchanger, hence Na
exchanger, hence Na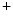 efflux in exchange of Ca
efflux in exchange of Ca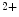 influx.11 In early, compensated maldaptive hypertrophy with preserved systolic contractility,
influx.11 In early, compensated maldaptive hypertrophy with preserved systolic contractility, 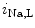 and APD remain unchanged. However, upon hypertension-induced heart failure with prolonged APD, the
and APD remain unchanged. However, upon hypertension-induced heart failure with prolonged APD, the 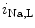 decay time is slower, but can be normalized by inhibition of Ca
decay time is slower, but can be normalized by inhibition of Ca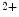 –calmodulin-dependent protein kinase CamK2 [398]. Increased CamK2 activity and subsequent Na
–calmodulin-dependent protein kinase CamK2 [398]. Increased CamK2 activity and subsequent Na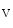 1.5 phosphorylation (Ser571) cause afterdepolarizations.
1.5 phosphorylation (Ser571) cause afterdepolarizations.
current () is indeed arrhythmogenic, as it induces an prolonged inward current favorable for EADs. Intracellular Na cycling is coupled with Ca homeostasis, as Na modulates the transport direction of the Na–Ca exchanger. Cellular Na overload as well as action potential prolongation stimulates the reverse mode of Na–Ca exchanger, hence Na efflux in exchange of Ca influx.11 In early, compensated maldaptive hypertrophy with preserved systolic contractility, and APD remain unchanged. However, upon hypertension-induced heart failure with prolonged APD, the decay time is slower, but can be normalized by inhibition of Ca–calmodulin-dependent protein kinase CamK2 [398]. Increased CamK2 activity and subsequent Na1.5 phosphorylation (Ser571) cause afterdepolarizations.Delayed Afterdepolarizations
Afterdepolarizations can result from aberrant calcium handling and provoke arrhythmogenesis. DADs during phase 3 of the action potential are caused by spontaneous Ca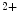 release from the sarcoplasmic reticulum. Calcium-induced DADs are modulated, but not generated by elevated sarcoplasmic reticulum Ca
release from the sarcoplasmic reticulum. Calcium-induced DADs are modulated, but not generated by elevated sarcoplasmic reticulum Ca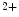 load. These afterdepolarizations require elevated calcium concentrations in the dyadic subspace and the cytosol that raise the open probability of ryanodin receptors [397].
load. These afterdepolarizations require elevated calcium concentrations in the dyadic subspace and the cytosol that raise the open probability of ryanodin receptors [397].
release from the sarcoplasmic reticulum. Calcium-induced DADs are modulated, but not generated by elevated sarcoplasmic reticulum Ca load. These afterdepolarizations require elevated calcium concentrations in the dyadic subspace and the cytosol that raise the open probability of ryanodin receptors [397].Spontaneous Ca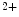 release triggers a premature action potential in failing cardiomyocytes that activates Na
release triggers a premature action potential in failing cardiomyocytes that activates Na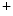 –Ca
–Ca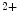 exchangers. The action potential in failing ventriculomyocytes is longer than in normal ones. The increased duration of the action potential is induced by enhanced activity of Na
exchangers. The action potential in failing ventriculomyocytes is longer than in normal ones. The increased duration of the action potential is induced by enhanced activity of Na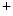 –Ca
–Ca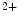 exchangers, slowed decay in Ca
exchangers, slowed decay in Ca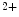 transient fluxes, and reduced activity of inwardly rectifier K
transient fluxes, and reduced activity of inwardly rectifier K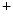 channels and Na
channels and Na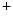 –K
–K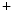 pumps.
pumps.
release triggers a premature action potential in failing cardiomyocytes that activates Na–Ca exchangers. The action potential in failing ventriculomyocytes is longer than in normal ones. The increased duration of the action potential is induced by enhanced activity of Na–Ca exchangers, slowed decay in Ca transient fluxes, and reduced activity of inwardly rectifier K channels and Na–K pumps.Metabolic Links to Electrical Dysfunction
The sequential oxidation of nutrients, fatty acids, and glucose leads to the common substrate for the tricarboxylic acid (TCA) cycle, acetylCoA (Vol. 6, Chap. 3. Cardiovascular Physiology). The latter is used to produce reducing equivalents NADH and FADH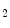 .
.
.Agent NADH feeds electrons to the electron transport chain. The redox reactions of the electron transport chain are coupled with proton translocation across the mitochondrial inner membrane. This H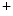 flux establishes an electrical potential at the mitochondrial membrane and pH gradient that both determine a proton-motive force. The latter is used by the mitochondrial ATP synthase to produce ATP, which is exported to the cytosol via the adenine nucleotide translocase (ANT).
flux establishes an electrical potential at the mitochondrial membrane and pH gradient that both determine a proton-motive force. The latter is used by the mitochondrial ATP synthase to produce ATP, which is exported to the cytosol via the adenine nucleotide translocase (ANT).
flux establishes an electrical potential at the mitochondrial membrane and pH gradient that both determine a proton-motive force. The latter is used by the mitochondrial ATP synthase to produce ATP, which is exported to the cytosol via the adenine nucleotide translocase (ANT).Although the electron transport chain efficiently utilizes O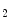 in the controlled oxidation of redox carriers, superoxide anion (O
in the controlled oxidation of redox carriers, superoxide anion (O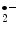 ) is an unavoidable by-product of oxidative phosphorylation.
) is an unavoidable by-product of oxidative phosphorylation.
in the controlled oxidation of redox carriers, superoxide anion (O) is an unavoidable by-product of oxidative phosphorylation.Mitochondria are involved in arrhythmogenesis. Mitochondria experience waves of membrane depolarization [399]. Periodic depolarizations of mitochondrial membranes are triggered by reactive oxygen species and propagated by ROS-induced ROS release (RIRR). Mitochondrial membrane potential oscillations involve ROS-sensitive mitochondrial inner membrane anion channels (IMAC) as well as slow depolarization waves related to mitochondrial permeability transition pore (mtPTP) opening.
ROS-induced ROS release causes oscillations of mitochondrial inner membrane potential, altered intracellular calcium dynamics via mitochondrial Ca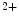 uniporter, and altered mitochondrial membrane potential due to the inner membrane anion channel, mitochondrial permeability transition pore, and mitochondrial ATP-sensitive K
uniporter, and altered mitochondrial membrane potential due to the inner membrane anion channel, mitochondrial permeability transition pore, and mitochondrial ATP-sensitive K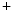 channels (
channels ( current), which shortens action potential.
current), which shortens action potential.
uniporter, and altered mitochondrial membrane potential due to the inner membrane anion channel, mitochondrial permeability transition pore, and mitochondrial ATP-sensitive K channels ( current), which shortens action potential.Adenosine triphosphate produced by mitochondria is mandatory for proper function of several cell determinants of electrical activity, such as ATPases SERCA and Na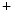 –K
–K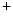 pump, as well as Ca
pump, as well as Ca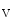 1.2 channel and sarcolemmal ATP-inactivated K
1.2 channel and sarcolemmal ATP-inactivated K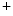 efflux channels (Vol. 6, Chap. 3. Cardiovascular Physiology). The latter increases heterogeneity of APD during metabolic stress, hence creating an arrhythmogenic substrate.
efflux channels (Vol. 6, Chap. 3. Cardiovascular Physiology). The latter increases heterogeneity of APD during metabolic stress, hence creating an arrhythmogenic substrate.
–K pump, as well as Ca1.2 channel and sarcolemmal ATP-inactivated K efflux channels (Vol. 6, Chap. 3. Cardiovascular Physiology). The latter increases heterogeneity of APD during metabolic stress, hence creating an arrhythmogenic substrate.Calcium ion transfer across the sarcolemma and sarcoplasmic reticulum during the excitation–contraction process is coupled with mitochondrial energetics and ROS metabolism (Table 5.1). Reactive oxygen species are produced by the electron transport chain, carried by the inner membrane anion channel (IMAC), and scavenged by superoxide dismutase and glutathione peroxidase (among others). This ROS-induced ROS release module and mitochondrial energetics are linked to cellular electrical activity via ATP-sensitive K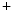 channel, which is activated when the ADP/ATP ratio increases [397].
channel, which is activated when the ADP/ATP ratio increases [397].
channel, which is activated when the ADP/ATP ratio increases [397].Table 5.1
Modules and components of the coupling between the excitation–contraction process, mitochondrial energetics, and ROS metabolism. (Source: [397])
Components | Elements or targets |
|---|---|
Mitochondrial energetics | |
TCA–ETC–ANT | NADH/FADH 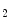 , ATP , ATP |
ATP | Myofilament |
Ion pumps (Na 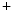 –K –K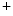 ATPase, PMCA, SERCA) ATPase, PMCA, SERCA) | |
Ion channels (K 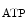 ) ) | |
Phosphocreatine circuit | mtCK–cyCK–smCK |
(ATP/ADP–Cr/Cr 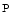 ) ) | |
ROS-induced ROS release | |
ETC | Superoxide (O 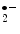 ) ) |
IMAC | Superoxide transfer |
Superoxide metabolism | SOD, GPx, GR |
Excitation–contraction process | |
Ca 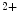 influx influx | Ca 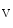 1.2 1.2 |
(entry and release) | RyR2 |
Mitochondrial Ca 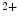 | mtCU (Ca 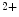 influx in matrix) influx in matrix) |
NCX (Ca 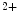 efflux from matrix) efflux from matrix) | |
ANT adenine nucleotide transporter, Cr creatine, Cr 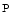 phosphorylated creatine, cy/mt/smCK cytosolic/mitochondrial/sarcomeric creatine kinase, ETC mitochondrial electron transport chain, GPx glutathione peroxidase, GR glutathione reductase, mtCU mitochondrial Ca
phosphorylated creatine, cy/mt/smCK cytosolic/mitochondrial/sarcomeric creatine kinase, ETC mitochondrial electron transport chain, GPx glutathione peroxidase, GR glutathione reductase, mtCU mitochondrial Ca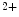 uniporter, PMCA plasma membrane Ca
uniporter, PMCA plasma membrane Ca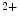 ATPase, SERCA sarco(endo)plasmic reticulum calcium ATPase, SOD superoxide dismutase, TCA tricarboxylic acid cycle
ATPase, SERCA sarco(endo)plasmic reticulum calcium ATPase, SOD superoxide dismutase, TCA tricarboxylic acid cycle
phosphorylated creatine, cy/mt/smCK cytosolic/mitochondrial/sarcomeric creatine kinase, ETC mitochondrial electron transport chain, GPx glutathione peroxidase, GR glutathione reductase, mtCU mitochondrial Ca uniporter, PMCA plasma membrane Ca ATPase, SERCA sarco(endo)plasmic reticulum calcium ATPase, SOD superoxide dismutase, TCA tricarboxylic acid cycleMyocardial ischemia and reperfusion (Sect. 8.8) provoke a metabolic stress, or ischemia–reperfusion injury, which culminates in electromechanical dysfunctions, such as ventricular tachycardia and fibrillation.
5.2.1.4 Reentry
Reentry requires a unidirectional block within a conducting path, the action potential traveling in the retrograde, but not in the direct direction, usually due to partial depolarization and suitable timing. The action potential travels into the common distal path, finds the nodal tissue excitable, runs across the block area in the reverse direction.
Reentry can occur either between the atria and ventricles, involving an accessory conduction path, or locally, within a small region of the atrium or ventricle. Reentrant circuits can involve a small (microreentry) or a large (macroreentry) cardiac region in a single chamber or in several chambers.
Since the 1980s, reentry was studied by many investigators in 2D and 3D, homogeneous, isotropic, excitable media [400–403]. An electromechanical model of a contracting excitable medium was recently proposed, the nonlinear excitation waves yielding contraction and the ensuing deformations exerting a feedback effect on the excitation properties of the medium [404]. A 3-variable FitzHugh–Nagumo-type excitation–tension finite difference model is coupled with a hyperelasticity finite element model.
5.2.2 Atrial Arrhythmias
The atrium contributes to about 25 % of the ventricular stroke volume. The initiation of the atrial contraction, similarly to ventricles, involves a nearly synchronous rise of intracellular Ca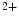 concentration supported by a well-developed transverse tubule network enriched in Ca
concentration supported by a well-developed transverse tubule network enriched in Ca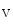 1.2 channels and Na
1.2 channels and Na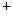 –Ca
–Ca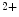 exchangers that are also concentrated along T-tubules. The former ion carrier accelerates changes in the amplitude of the systolic Ca
exchangers that are also concentrated along T-tubules. The former ion carrier accelerates changes in the amplitude of the systolic Ca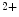 transient during inotropic stimuli and the latter lowers the diastolic cytosolic Ca
transient during inotropic stimuli and the latter lowers the diastolic cytosolic Ca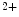 concentration.
concentration.
concentration supported by a well-developed transverse tubule network enriched in Ca1.2 channels and Na–Ca exchangers that are also concentrated along T-tubules. The former ion carrier accelerates changes in the amplitude of the systolic Ca transient during inotropic stimuli and the latter lowers the diastolic cytosolic Ca concentration.Apposed Ca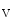 1.2 and RyR channels that form couplons localize close to NCX antiporters, hence favoring a positive lusitropic effect, as they lower the cytosolic Ca
1.2 and RyR channels that form couplons localize close to NCX antiporters, hence favoring a positive lusitropic effect, as they lower the cytosolic Ca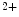 level. However, the resulting inward depolarizing current can be proarrhythmogenic [405].
level. However, the resulting inward depolarizing current can be proarrhythmogenic [405].
1.2 and RyR channels that form couplons localize close to NCX antiporters, hence favoring a positive lusitropic effect, as they lower the cytosolic Ca level. However, the resulting inward depolarizing current can be proarrhythmogenic [405].An excessive RyR phosphorylation by PKA or CamK causes Ca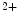 leakage from the sarcoplasmic reticulum. Subsequent Ca
leakage from the sarcoplasmic reticulum. Subsequent Ca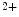 efflux through NCX carriers depolarizes the myocyte, thereby initiating and maintaining arrhythmia.
efflux through NCX carriers depolarizes the myocyte, thereby initiating and maintaining arrhythmia.
leakage from the sarcoplasmic reticulum. Subsequent Ca efflux through NCX carriers depolarizes the myocyte, thereby initiating and maintaining arrhythmia.In ventriculomyocytes, the T-tubular orientation is predominantly perpendicular to the long myocyte axis at each Z line with branching between adjacent tubules. Calcium sparks occur predominantly at Z lines along the length of T-tubules.
In atriomyocytes, the T-tubular orientation is mainly axial along the long myocyte axis and less organized than in ventriculomyocytes. In addition, the density of Ca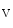 1.2–RyR couplons is smaller. Furthermore, regional differences exist between left and right atria, appendage and free wall, among other territories [405]. Narrow myocytes have a smaller density of T-tubules than wide myocytes. Hence, the spatial and temporal properties of systolic Ca
1.2–RyR couplons is smaller. Furthermore, regional differences exist between left and right atria, appendage and free wall, among other territories [405]. Narrow myocytes have a smaller density of T-tubules than wide myocytes. Hence, the spatial and temporal properties of systolic Ca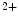 transients differ between the two myocyte types. However, T-tubules in atriomyocytes create simultaneously Ca
transients differ between the two myocyte types. However, T-tubules in atriomyocytes create simultaneously Ca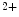 transient at the myocyte edge and center [405].
transient at the myocyte edge and center [405].
1.2–RyR couplons is smaller. Furthermore, regional differences exist between left and right atria, appendage and free wall, among other territories [405]. Narrow myocytes have a smaller density of T-tubules than wide myocytes. Hence, the spatial and temporal properties of systolic Ca transients differ between the two myocyte types. However, T-tubules in atriomyocytes create simultaneously Ca transient at the myocyte edge and center [405].In addition, inositol trisphosphate receptors activated by IP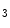 generated upon stimulation by endothelin-1, angiotensin-2, noradrenaline that targets α1-adrenoceptor, and acetylcholine that bins to muscarinic M
generated upon stimulation by endothelin-1, angiotensin-2, noradrenaline that targets α1-adrenoceptor, and acetylcholine that bins to muscarinic M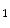 and M
and M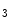 receptors contribute to Ca
receptors contribute to Ca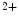 release from IP
release from IP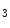 -sensitive Ca
-sensitive Ca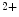 stores. This Ca
stores. This Ca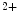 influx can stimulate RyR-dependent Ca
influx can stimulate RyR-dependent Ca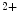 release.
release.
generated upon stimulation by endothelin-1, angiotensin-2, noradrenaline that targets α1-adrenoceptor, and acetylcholine that bins to muscarinic M and M receptors contribute to Ca release from IP-sensitive Ca stores. This Ca influx can stimulate RyR-dependent Ca release.Atriomyocyte contraction relies on Ca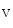 1.2 and RyR channels. Atriomyocyte relaxation depends on SERCA pump, Na
1.2 and RyR channels. Atriomyocyte relaxation depends on SERCA pump, Na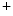 –Ca
–Ca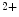 exchanger, and, to a lesser extent, plasmalemmal PMCA pump. The difference of functioning between atrial and ventricular myocytes is related to the control of systolic Ca
exchanger, and, to a lesser extent, plasmalemmal PMCA pump. The difference of functioning between atrial and ventricular myocytes is related to the control of systolic Ca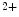 transients. In the rat, atriomyocytes devoid of T-tubules are smaller and have a reduced Ca
transients. In the rat, atriomyocytes devoid of T-tubules are smaller and have a reduced Ca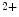 transient amplitude, higher rate of [Ca
transient amplitude, higher rate of [Ca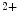 ]
]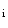 decay due to greater SERCA activity, larger Ca
decay due to greater SERCA activity, larger Ca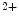 buffering capacity associated with a higher SERCA/phospholamban ratio, and elevated Ca
buffering capacity associated with a higher SERCA/phospholamban ratio, and elevated Ca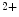 store content than ventriculomyocytes [405].
store content than ventriculomyocytes [405].
1.2 and RyR channels. Atriomyocyte relaxation depends on SERCA pump, Na–Ca exchanger, and, to a lesser extent, plasmalemmal PMCA pump. The difference of functioning between atrial and ventricular myocytes is related to the control of systolic Ca transients. In the rat, atriomyocytes devoid of T-tubules are smaller and have a reduced Ca transient amplitude, higher rate of [Ca] decay due to greater SERCA activity, larger Ca buffering capacity associated with a higher SERCA/phospholamban ratio, and elevated Ca store content than ventriculomyocytes [405].Anchoring proteins, such as junctophilin-2, amphiphysin-2, and telothionin, connect to Z-disc and cytoskeleton constituents and partcipate in the formation of T-tubules in cardiomyocytes and trafficking of ion channels to the T-tubule membrane [405]. Altered expression of these proteins leads to a T-tubule disorganization or loss.
In atria that are the site of common cardiac arrhythmia, the T-tubular network can be remodeled with disorganization and loss in heart failure and atrial fibrillation. In atrial fibrillation, the systolic rise of Ca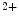 level is considerably less uniform [405].
level is considerably less uniform [405].
level is considerably less uniform [405].Certain regions of the atria such as pulmonary vein cardiomyocytes, in which intracellular Ca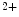 regulation differs from that of atriomyocytes, can be involved in the genesis of atrial fibrillation. An early pulmonary vein myocardium formation under the control of the paired-like homeodomain transcription factor Pitx2c is strongly associated with future occurrence of atrial fibrillation [405].
regulation differs from that of atriomyocytes, can be involved in the genesis of atrial fibrillation. An early pulmonary vein myocardium formation under the control of the paired-like homeodomain transcription factor Pitx2c is strongly associated with future occurrence of atrial fibrillation [405].
regulation differs from that of atriomyocytes, can be involved in the genesis of atrial fibrillation. An early pulmonary vein myocardium formation under the control of the paired-like homeodomain transcription factor Pitx2c is strongly associated with future occurrence of atrial fibrillation [405].5.2.2.1 Sinusal Bradycardia and Tachycardia
A sinus rhythm12 with normal, evenly spaced complexes and a frequency lower than 1 Hz (60/mn) is called sinus bradycardia.
A sinus rhythm with a frequency higher than 1.67 Hz (100/mn) is termed sinus tachycardia.
Irregular sinus rhythms with the longest RR interval (exceeding the usual value by 160 ms) are called sinus arrhythmias.
5.2.2.2 Sinus Node Dysfunction
Sinusal node dysfunction (SND), or sick sinus syndrome (SSS), and atrioventricular node block are common causes of bradyarrhythmias that often necessitate permanent pacemaker placement. Sinus node dysfunction causes sinus bradycardia, sinus arrest, exit block, combinations of sinoatrial and atrioventricular nodal defects, and atrial tachyarrhythmias.
Sinusal node disease, a common indication for permanent cardiac pacing, results from disordered impulse generation within the sinusal node or impaired conduction of the impulse to the surrounding atrial wall, thereby engendering bradycardia. Its natural evolution is marked by recurrent syncope, heart failure, stroke, and atrial fibrillation.
Sinusal node disease is associated with diffuse atrial remodeling characterized by structural changes, conduction anomalies, and increased right atrial refractoriness. The sinusal pacemaker activity varies with a loss of the normal multicentric pattern of activation, caudal shift of the pacemaker, and abnormal conduction around lines of conduction block [374].
Whereas human atrial and ventricular arrhythmias can result from many defective ion channel genes, genetic and molecular mechanisms responsible for sinus node dysfunction are associated with ankyrin-B locus ANK2 [406]. Ankyrin-B serves in membrane organization of sinoatrial node cell channels and transporters and hence in cardiac pacing. Variants of the ANK2 gene is also responsible for congenital type-4 LQTS.
5.2.2.3 Ectopic Pacemaker
The origin of atrial contractions can localize elsewhere in the atria other than the sinusal node. If the ectopic pacemaker localizes close to the atrioventricular node, the atrial depolarization occurs in the opposite direction with respect to the normal one, with inversion of P-wave polarity. Any ectopic pacemaker located in the junction between the atria and ventricles generates a junctional rhythm with slow cardiac frequency (40–55/mn, i.e., 0.66–0.92 Hz), normal QRS complexes, and vanishing P waves.
5.2.2.4 Paroxysmal Atrial Tachycardia
Paroxysmal atrial tachycardia, with a frequency of 160–220/mn (2.67–3.67 Hz), results from reentrant activation. P waves are immediately followed by QRS complexes.
5.2.2.5 Atrial Flutter
Atrial flutter occurs when the atria contract two or three times for each ventricular contraction. Atrial flutter is characterized by TP interval disappearance.
5.2.2.6 Atrial Fibrillation
Atrial fibrillation (AF), the most common sustained cardiac arrhythmia, causes a rapid and irregular atrial activity. Atriomyocytes fire at a frequency ranging from 6.7 to 10 Hz (rate 400–600/mn). The atria quiver rather than truly contract.
Clinical Features
Obesity is a risk factor, especially when it coexists with hypertension, diabetes, and sleep apnea. Atrial fibrillation causes palpitations and dizziness as well as complications, such as stroke and heart failure.
Atrial fibrillations are classified into three main types:
1.
Paroxysmal, intermittent, usually self-terminating
2.
Persistent
3.
Permanent atrial fibrillations
Main Electrophysiological Features
The ventricular frequency is also high (typically 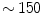 pulses/mn, i.e.,
pulses/mn, i.e., 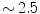 Hz) and irregular with normal QRS complexes. The ventricular beating rate is dictated by the interactions between the atrial rate and the filtering function of the atrioventricular node.
Hz) and irregular with normal QRS complexes. The ventricular beating rate is dictated by the interactions between the atrial rate and the filtering function of the atrioventricular node.
pulses/mn, i.e., Hz) and irregular with normal QRS complexes. The ventricular beating rate is dictated by the interactions between the atrial rate and the filtering function of the atrioventricular node.The multiple wavelet hypothesis relies on the existence of randomly propagating waves with intermittent blockades and annihilation and regeneration of waves responsible for a self-sustained, spatiotemporally complex patterns of excitation lacking a hierarchical organization. However, wave propagation during atrial fibrillation is not random and reentry is a major mechanism of fibrillation.
The occurrence and persistence of atrial fibrillation require the combined effect of a trigger, substrate, and activation of the autonomous nervous system [68]. The trigger consists of focal spontaneous electrical activity (focal drivers) localized to preferential sites, mainly the pulmonary vein. The substrate is related to structural and functional alterations of the atrial myocardium that engender the shortening of atrial refractoriness, formation of reentry circuits, and local conduction blocks. The reentrant activity maintains the arrhythmia.
Anatomical Background
The anatomy of atria with pectinate muscles, the crista terminalis, vein orifices, and atrioventricular rings is a favorable milieu for vortices of electrochemical waves (reentrant rotors or spiral waves) [407]. The crista terminalis, pectinate muscles, and Bachman bundle have greater anisotropy ratios and conduction speeds than the remaining atrial myocardium [408]. Pulmonary veins and the ligament of Marshall act as high-frequency sources [407]. Hence, regions of the posterior left atrium can operate as AF drivers.
The Bachmann bundle and inferoposterior interatrial pathway that underlies the coronary sinus as well as the superior and inferior aspects of the fossa ovalis are routes of interatrial electrical communication that can mediate fibrillatory conduction [407].
At least some cases of paroxysmal and chronic atrial fibrillation result from uninterrupted periodic activity of reentrant sites. A single or few high-frequency reentrant sources can localize to the left atrium [407]. Sources of high-frequency periodic activity, dispersion of local activation, and stable, self-sustained rotors can exist in the atria [409].
Reentrant rotors in the left atrium are faster than those in the right atrium [410]. The acetylcholine-modulated potassium inward rectifier current (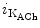 ) is higher in the left atrium than in the right atrium. The sensitivity of LA rotors to ACh is then greater than that of RA rotors.
) is higher in the left atrium than in the right atrium. The sensitivity of LA rotors to ACh is then greater than that of RA rotors.
) is higher in the left atrium than in the right atrium. The sensitivity of LA rotors to ACh is then greater than that of RA rotors.In some paroxysmal AF patients, rapid impulses triggered by an ectopic pacemaker propagate promptly as succeeding wave fronts from a pulmonary vein into the left atrium to encounter heterogeneously recovered tissue [407]. When conditions of heterogeneity are appropriate, the wave fronts brake and initiate two counter-rotating vortices that may be stable. On the other hand, a single rotor persists and engenders wave fronts at an exceedingly high frequency. The AF initiation and maintenance may then depend on the formation of relatively sustained rotors in the left atrium that generate fibrillatory waves.
Histological Background
Autonomic innervation of the heart encompasses the extrinsic (ECANS) and intrinsic (ICANS) cardiac autonomic nervous system. The letter consists of a neural network with ganglionated plexi concentrated within epicardial fat pads and interconnecting ganglia and axons.13 The ganglionated plexi intervene in the initiation and maintenance of atrial fibrillation.
The Na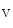 1.8 channel encoded by the SCN10A gene is highly expressed in thin sensory neurons of dorsal root ganglia. It also lodges in the intrinsic cardiac ganglia. It is involved in atrial fibrillation. In cardiac ganglionated plexi, blockade of Na
1.8 channel encoded by the SCN10A gene is highly expressed in thin sensory neurons of dorsal root ganglia. It also lodges in the intrinsic cardiac ganglia. It is involved in atrial fibrillation. In cardiac ganglionated plexi, blockade of Na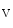 1.8 suppresses the effects of vagus nerve stimulation on cardiac conduction [411]. This effect results most likely from inhibition of the parasympathetic ganglionic neurotransmission in the ICANS.
1.8 suppresses the effects of vagus nerve stimulation on cardiac conduction [411]. This effect results most likely from inhibition of the parasympathetic ganglionic neurotransmission in the ICANS.
1.8 channel encoded by the SCN10A gene is highly expressed in thin sensory neurons of dorsal root ganglia. It also lodges in the intrinsic cardiac ganglia. It is involved in atrial fibrillation. In cardiac ganglionated plexi, blockade of Na1.8 suppresses the effects of vagus nerve stimulation on cardiac conduction [411]. This effect results most likely from inhibition of the parasympathetic ganglionic neurotransmission in the ICANS.The electrical properties of atriomyocytes can be altered. In the myocardium, impulse propagation depends on the excitability of myocytes, discharge transmission between adjacent myocytes, and the three-dimensional arrangement of myocytes. The anisotropic electrochemical wave propagation follows myofibers with discontinuity that may be sources of reentrant rotors.
Nodal cells of the conduction paths and myocytes that are characterized by their orientation are organized in bundles,14 in atria and ventricles (size  [100 μm–
[100 μm– [100 mm) [408]. Aligned tracts of nodal tissue such as Purkinje fibers are in fact 3D arrays of cells.
[100 mm) [408]. Aligned tracts of nodal tissue such as Purkinje fibers are in fact 3D arrays of cells.
[100 μm–[100 mm) [408]. Aligned tracts of nodal tissue such as Purkinje fibers are in fact 3D arrays of cells.Electrochemical wave propagates preferentially along the myofiber axis. The action potential transversal spreading rate depends on the extent of lateral coupling between adjacent myocytes.
In addition, fibroblasts and myofibroblasts may influence cardiomyocyte electrophysiology via juxtacrine interactions (bidirectional coupling via gap junctions) and modulation of action potential transmission. Cardiofibroblasts are also arranged in a network that surrounds cardiomyocytes. Myocardial injury engenders myofibroblasts (absent in the normal myocardium) that have greater proliferation and collagen production rates than fibroblasts. Cardiac fibroblasts and myofibroblasts possess ion carriers, among which ion channels that carry inwardly rectifying and various voltage-dependent outward K currents. Their resting transmembrane potential is less negative than that of cardiomyocytes [408]. The fibroblast (myofibroblast) network may then act as a current source for adjacent inactive cardiomyocytes and as a sink during activation. Myofibroblast density-dependent reductions in cardiomyocyte resting transmembrane potential, conduction velocity slowing, and propagation across fibroblast barriers can be observed.
currents. Their resting transmembrane potential is less negative than that of cardiomyocytes [408]. The fibroblast (myofibroblast) network may then act as a current source for adjacent inactive cardiomyocytes and as a sink during activation. Myofibroblast density-dependent reductions in cardiomyocyte resting transmembrane potential, conduction velocity slowing, and propagation across fibroblast barriers can be observed.
currents. Their resting transmembrane potential is less negative than that of cardiomyocytes [408]. The fibroblast (myofibroblast) network may then act as a current source for adjacent inactive cardiomyocytes and as a sink during activation. Myofibroblast density-dependent reductions in cardiomyocyte resting transmembrane potential, conduction velocity slowing, and propagation across fibroblast barriers can be observed.Adverse atrial structural remodeling such as diffuse fibrosis causes electrophysiological disturbances due to conduction abnormalities. Atrial fibrillation is associated with cardiac and pulmonary diseases. Several cardiac disorders predispose to atrial fibrillations, such as coronary artery diseases, pericarditis, valvular diseases, congenital heart diseases, congestive heart failure, thyrotoxic heart disease, sleep apnea, and hypertension. Idiopathic atrial fibrillation can be associated with mutations in the gene encoding connexin-40.
Impulse propagation in the cardiac wall can be modeled according to the one-dimensional continuous cable theory. This framework is useful to point out the role of current source-to-sink matching responsible for conduction slowing and unidirectional block, but it cannot represent entirely electrical properties of 3D networks of myocytes. Discharge propagation in discontinuous 1D strands of myocytes coupled by discrete resistive pathways differs from the prediction of the cable theory.
Impulse transmission between cells can follow the ephaptic15 transmission, that is, the passage of an action potential from the prejunctional cell to the postjunctional cell through their membranes across an electrochemical synapse [ephapse] in a saltatory fashion driven by the electrical potential in the cleft between two apposed cells associated with the exchange of ions between the cells. Sodium channels that are concentrated around intercalated discs support the ephaptic transmission.
Discontinuous arrangement of myocytes and the connective tissue can engender current source-to-sink mismatch, spatiotemporal distribution of refractoriness, and rate-sensitive electric instability, which contribute to the initiation and maintenance of reentry [408]. Electrochemical dysfunction and structural remodeling (fibrosis) can generate ectopic activation and local propagation delays that support reentry.
Biochemical Background
The activated renin–angiotensin–aldosterone axis, platelet-derived and connective tissue growth factor, and local inflammation secondary to endothelial dysfunction and thrombus formation favor fibrosis. The abundance of atrial fatty deposits is related to the risk and severity of atrial fibrillation.
The refractory period depends on the APD. Smaller inward currents ( ) and higher outward currents (K
) and higher outward currents (K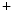 currents [
currents [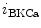 ]) reduce the refractory period and promote atrial fibrillation [412].
]) reduce the refractory period and promote atrial fibrillation [412].
) and higher outward currents (K currents []) reduce the refractory period and promote atrial fibrillation [412].Regulation by phosphatases of Ca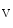 1.2 can be impaired. Transcript levels of PP1 and PP2 decrease, the PP1 level is not altered, but the PP2 level increases in AF patients. The phosphatase PP1 is the major subtype of protein Ser/Thr phosphatases in the heart [413]. The inhibitor PP1
1.2 can be impaired. Transcript levels of PP1 and PP2 decrease, the PP1 level is not altered, but the PP2 level increases in AF patients. The phosphatase PP1 is the major subtype of protein Ser/Thr phosphatases in the heart [413]. The inhibitor PP1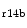 is activated by PKA to prevent PP1 target dephosphorylation, thereby enabling amplification of β-adrenoceptor signaling. The activity of both PP1 and PP2 rises in patients with chronic AF, but in a heterogeneous manner according to the subcellular compartment. Phosphorylation of myosin-binding protein-C (Ser282) by PKA lowers, that of troponin-I is preserved, and that of phospholamban (Ser16 and Thr17) augments.
is activated by PKA to prevent PP1 target dephosphorylation, thereby enabling amplification of β-adrenoceptor signaling. The activity of both PP1 and PP2 rises in patients with chronic AF, but in a heterogeneous manner according to the subcellular compartment. Phosphorylation of myosin-binding protein-C (Ser282) by PKA lowers, that of troponin-I is preserved, and that of phospholamban (Ser16 and Thr17) augments.
1.2 can be impaired. Transcript levels of PP1 and PP2 decrease, the PP1 level is not altered, but the PP2 level increases in AF patients. The phosphatase PP1 is the major subtype of protein Ser/Thr phosphatases in the heart [413]. The inhibitor PP1 is activated by PKA to prevent PP1 target dephosphorylation, thereby enabling amplification of β-adrenoceptor signaling. The activity of both PP1 and PP2 rises in patients with chronic AF, but in a heterogeneous manner according to the subcellular compartment. Phosphorylation of myosin-binding protein-C (Ser282) by PKA lowers, that of troponin-I is preserved, and that of phospholamban (Ser16 and Thr17) augments.Phosphorylation of the ryanodine receptor RyR2 by protein kinase-A (Ser2808) and Ca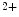 –calmodulin-dependent protein kinase CamK2 (Ser2814) is antagonized by the protein phosphatases PP1 and PP2. Impaired local regulation of protein phosphatase PP1 leads to hyperphosphorylation (hyperactivity) of RyR2, thereby favoring atrial fibrillation [414]. PP1 dephosphorylates RyR2 that was phosphorylated by the CamK2 kinase. Spinophilin, or PP1
–calmodulin-dependent protein kinase CamK2 (Ser2814) is antagonized by the protein phosphatases PP1 and PP2. Impaired local regulation of protein phosphatase PP1 leads to hyperphosphorylation (hyperactivity) of RyR2, thereby favoring atrial fibrillation [414]. PP1 dephosphorylates RyR2 that was phosphorylated by the CamK2 kinase. Spinophilin, or PP1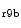 , assists PP1 in targeting RyR2. Upon ablation of spinophilin, the interaction between PP1 and RyR2 lowers (64 %); RyR2 phosphorylation rises (43 % at Ser2814), but remains unchanged at Ser2808; and open RyR2 probability and subsequently Ca
, assists PP1 in targeting RyR2. Upon ablation of spinophilin, the interaction between PP1 and RyR2 lowers (64 %); RyR2 phosphorylation rises (43 % at Ser2814), but remains unchanged at Ser2808; and open RyR2 probability and subsequently Ca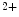 spark frequency augments.
spark frequency augments.
–calmodulin-dependent protein kinase CamK2 (Ser2814) is antagonized by the protein phosphatases PP1 and PP2. Impaired local regulation of protein phosphatase PP1 leads to hyperphosphorylation (hyperactivity) of RyR2, thereby favoring atrial fibrillation [414]. PP1 dephosphorylates RyR2 that was phosphorylated by the CamK2 kinase. Spinophilin, or PP1, assists PP1 in targeting RyR2. Upon ablation of spinophilin, the interaction between PP1 and RyR2 lowers (64 %); RyR2 phosphorylation rises (43 % at Ser2814), but remains unchanged at Ser2808; and open RyR2 probability and subsequently Ca spark frequency augments.Transcription factors contribute to cardiac arrhythmias. They can create a proarrhythmogenic ground in the pulmonary veins and atrium. They are implicated in atrial remodeling linked to atrial fibrillation, such as TSC22, TcEb, EGR2, GTF2h2, and Fos, as well as transcription factor-related proteins, such as cardiac ankyrin repeat protein (CARP) and Four and a half LIM domain-containing protein FHL1 [415]. Mutations in genes that encode transcription factors, such as TBx5,16 GATA4 to GATA6,17 and NKx2-5 are responsible for familial forms of atrial fibrillation (Table 5.2; [415]).
Table 5.2
Transcription factors implicated in atrial fibrillation. (Source: [415])
Transcription factors | Targets |
|---|---|
GATA4 | NPPa |
GATA5 | NPPa |
GATA6 | NPPa |
NKx2-5 | MinK, Na 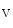 1.5, Cx40, NPPa 1.5, Cx40, NPPa |
Pitx2 | HCN4, NPPa |
TBx5 | K 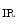 2.1, Na 2.1, Na 1.5, Cx40, NPPa 1.5, Cx40, NPPa |
Cx40 connexin-40 encode by the GJA5 gene, HCN hyper- polarization-activated, cyclic nucleotide-gated K channel, NPPa natriuretic peptide precursor-A
channel, NPPa natriuretic peptide precursor-A
channel, NPPa natriuretic peptide precursor-AThe TBX5 gene mutation (G125R) is associated with a gain of function and increased NPPA (natriuretic peptide precursor-A), CX40 (connexin-40), and KCNJ2 (K 2.1) gene expression [415]. Mutations in the Gata4 gene (G16C, H28D, Y38D, S70T, P103A, and S160T), Gata5 gene (Y138F, W200G, and C210G), and Gata6 gene (Y235S) are associated with a loss of function. The NKX2-5 gene mutation (T768A) diminishes the ability of NKx2-5 to dimerize, but not of transactivating the NPPA gene.
2.1) gene expression [415]. Mutations in the Gata4 gene (G16C, H28D, Y38D, S70T, P103A, and S160T), Gata5 gene (Y138F, W200G, and C210G), and Gata6 gene (Y235S) are associated with a loss of function. The NKX2-5 gene mutation (T768A) diminishes the ability of NKx2-5 to dimerize, but not of transactivating the NPPA gene.
2.1) gene expression [415]. Mutations in the Gata4 gene (G16C, H28D, Y38D, S70T, P103A, and S160T), Gata5 gene (Y138F, W200G, and C210G), and Gata6 gene (Y235S) are associated with a loss of function. The NKX2-5 gene mutation (T768A) diminishes the ability of NKx2-5 to dimerize, but not of transactivating the NPPA gene.In addition, the risk single nucleotide polymorphisms for atrial fibrillation localize to the vicinity of transcription factor PITX2, ZFHX3, and PRRX1 genes [415].
Rapidly Discharging Ectopic Sites and Reentries
Atrial fibrillation is initiated and maintained by at least one ectopic focus with rapid activity associated with a single small reentry circuit or multiple reentries [412]. Reentrant loops in the atrial myocardium are triggered by rapid discharges of ectopic foci, commonly in sleeves of atrial tissue located within the pulmonary veins. Increased activity of Na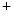 –Ca
–Ca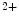 exchangers promotes afterdepolarization-related atrial ectopic firing.
exchangers promotes afterdepolarization-related atrial ectopic firing.
–Ca exchangers promotes afterdepolarization-related atrial ectopic firing.Atrial fibrillation is launched by a substrate available for reentries generated by a trigger, usually spontaneous focal ectopic discharges. Focal ectopic discharges include EADs and DADs (Table 5.3). The size of reentry circuits is determined by the wavelength, that is, the product of refractory period by conduction velocity; the smaller is the wavelength, the shorter the APD, the smaller the reentry circuits and the higher their number, and the more sustained the atrial fibrillation. When the number of circuits in the atria is small, reentry is unstable and atrial fibrillation self-terminates.
Table 5.3
Mechanisms of atrial fibrillation (Source: [385]). During the systole, Ca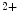 entry through Ca
entry through Ca 1.2a triggers systolic Ca
1.2a triggers systolic Ca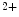 release from the sarcoplasmic reticulum into the cytosol through ryanodine receptor RyR2. During diastole, normal resting cytosolic Ca
release from the sarcoplasmic reticulum into the cytosol through ryanodine receptor RyR2. During diastole, normal resting cytosolic Ca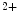 concentration is restored by Ca
concentration is restored by Ca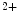 export, especially reuptake by the sarcoplasmic reticulum Ca
export, especially reuptake by the sarcoplasmic reticulum Ca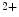 ATPase SERCA2a and extrusion into the extracellular space via Na
ATPase SERCA2a and extrusion into the extracellular space via Na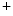 –Ca
–Ca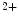 exchange (NCX). In delayed afterdepolarization (DAD), abnormal diastolic Ca
exchange (NCX). In delayed afterdepolarization (DAD), abnormal diastolic Ca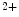 release through RyR2s because of excess sarcoplasmic reticulum Ca
release through RyR2s because of excess sarcoplasmic reticulum Ca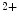 load reaching release threshold or enhanced RyR Ca
load reaching release threshold or enhanced RyR Ca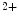 sensitivity causes a transient inward (subscript ti) current through NCX (
sensitivity causes a transient inward (subscript ti) current through NCX (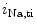 ). In early afterdepolarizations (EADs), action potential prolongation enables
). In early afterdepolarizations (EADs), action potential prolongation enables 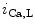 current to depolarize the cell from the plateau phase
current to depolarize the cell from the plateau phase
entry through Ca1.2a triggers systolic Ca release from the sarcoplasmic reticulum into the cytosol through ryanodine receptor RyR2. During diastole, normal resting cytosolic Ca concentration is restored by Ca export, especially reuptake by the sarcoplasmic reticulum Ca ATPase SERCA2a and extrusion into the extracellular space via Na–Ca exchange (NCX). In delayed afterdepolarization (DAD), abnormal diastolic Ca release through RyR2s because of excess sarcoplasmic reticulum Ca load reaching release threshold or enhanced RyR Ca sensitivity causes a transient inward (subscript ti) current through NCX (). In early afterdepolarizations (EADs), action potential prolongation enables current to depolarize the cell from the plateau phaseTrigger | Action potential phase | Involved ion carrier(s) |
|---|---|---|
DAD | 4 (rest) | Na  –Ca –Ca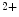 exchanger exchanger( 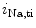 current) current) |
RyR2 (abnormal activity) | ||
Effect: postrefractory period depolarization | ||
EAD | 2 (plateau end) | Ca 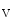 1.2 1.2 |
( 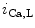 current) current) | ||
K 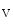 1.5 1.5 | ||
( 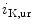 current) current) | ||
K  | ||
Na 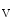 1.5 channel 1.5 channel | ||
( 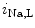 current) current) | ||
3 (final repolarization) | Na 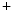 –Ca –Ca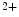 exchange current exchange current | |
( 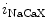 current) current) | ||
Origin: long action potential duration | ||
Effect: surrounding myocardium depolarization | ||
Early Afterdepolarizations
EADs precede full repolarization, that is, they happen at the end of plateau or early in the final phase of repolarization. EADs occur when the action potential is excessively prolonged.
Long duration of action potentials can result from [385]: (1) increased inward 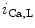 current through Ca
current through Ca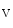 1.2 channel; (2) late
1.2 channel; (2) late 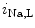 current through Na
current through Na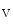 1.5; and/or (3) reduced K
1.5; and/or (3) reduced K currents. Recovery from inactivation of the Ca
currents. Recovery from inactivation of the Ca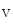 1.2 channel, enhanced inward Na
1.2 channel, enhanced inward Na flux through the Na
flux through the Na –Ca
–Ca exchanger, and phosphorylation by CamK2 of Ca
exchanger, and phosphorylation by CamK2 of Ca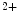 channels contribute to EAD occurrence.
channels contribute to EAD occurrence.
current through Ca1.2 channel; (2) late current through Na1.5; and/or (3) reduced K currents. Recovery from inactivation of the Ca1.2 channel, enhanced inward Na flux through the Na–Ca exchanger, and phosphorylation by CamK2 of Ca channels contribute to EAD occurrence.Na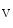 1.5 channel is primarily responsible for action potential initiation and propagation in the heart. Its fast inactivation occurs within ms after membrane depolarization onset. It determines APD. Inherited mutations in the SCN5A gene that encodes Na
1.5 channel is primarily responsible for action potential initiation and propagation in the heart. Its fast inactivation occurs within ms after membrane depolarization onset. It determines APD. Inherited mutations in the SCN5A gene that encodes Na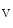 1.5 can disrupt fast inactivation and promote persistent channel activity that can prolong APD and cause the congenital long-QT syndrome (LQT3), Brugada syndrome, and isolated conduction disease [416].
1.5 can disrupt fast inactivation and promote persistent channel activity that can prolong APD and cause the congenital long-QT syndrome (LQT3), Brugada syndrome, and isolated conduction disease [416].
1.5 channel is primarily responsible for action potential initiation and propagation in the heart. Its fast inactivation occurs within ms after membrane depolarization onset. It determines APD. Inherited mutations in the SCN5A gene that encodes Na1.5 can disrupt fast inactivation and promote persistent channel activity that can prolong APD and cause the congenital long-QT syndrome (LQT3), Brugada syndrome, and isolated conduction disease [416].Loss-of-function mutations of genes that encode K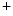 channels, either those linked to LQTS or those responsible for the atrial ultrarapid delayed rectifier K
channels, either those linked to LQTS or those responsible for the atrial ultrarapid delayed rectifier K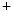 current (
current (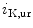 ), i.e., K
), i.e., K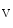 1.5 channel, are associated with atrial fibrillation due to ectopic site activity related to EADs [385].
1.5 channel, are associated with atrial fibrillation due to ectopic site activity related to EADs [385].
channels, either those linked to LQTS or those responsible for the atrial ultrarapid delayed rectifier K current (), i.e., K1.5 channel, are associated with atrial fibrillation due to ectopic site activity related to EADs [385].Atrial fibrillation relying on EADs can also be induced by gain-of-function mutations of genes that encode ion channels carrying 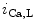 or
or 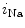 (Na
(Na 1.5) current, which also cause LQTS [385].
1.5) current, which also cause LQTS [385].
or (Na1.5) current, which also cause LQTS [385].Long APD enables the Ca 1.2 channel to recover and depolarize the cell by Ca
1.2 channel to recover and depolarize the cell by Ca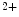 entry. EADs cause ectopic firing by depolarizing surrounding myocardium to excitation threshold.
entry. EADs cause ectopic firing by depolarizing surrounding myocardium to excitation threshold.
1.2 channel to recover and depolarize the cell by Ca entry. EADs cause ectopic firing by depolarizing surrounding myocardium to excitation threshold.Delayed Afterdepolarizations
DADs occur after full repolarization. They are caused by Na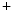 –Ca
–Ca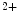 exchange current (
exchange current (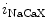 or
or 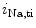 : transient inward Na
: transient inward Na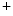 current) generated by a transient diastolic rise in cytosolic Ca
current) generated by a transient diastolic rise in cytosolic Ca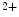 concentration due to abnormal RyR2 activity. Additional Ca
concentration due to abnormal RyR2 activity. Additional Ca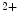 ions are exchanged for extracellular Na
ions are exchanged for extracellular Na ion in a 1:3 ionic ratio, creating a net inward movement of positive ions [385].
ion in a 1:3 ionic ratio, creating a net inward movement of positive ions [385].
–Ca exchange current ( or : transient inward Na current) generated by a transient diastolic rise in cytosolic Ca concentration due to abnormal RyR2 activity. Additional Ca ions are exchanged for extracellular Na ion in a 1:3 ionic ratio, creating a net inward movement of positive ions [385].Arrhythmogenic diastolic Ca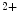 leak through RyR2s can result from: (1) excess sarcoplasmic reticulum Ca
leak through RyR2s can result from: (1) excess sarcoplasmic reticulum Ca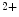 load reaching release threshold or (2) enhanced RyR Ca
load reaching release threshold or (2) enhanced RyR Ca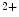 sensitivity. Hyperphosphorylation of phospholamban, a SERCA2a inhibitor, relieves SERCA inhibition, hence increasing sarcoplasmic reticulum Ca
sensitivity. Hyperphosphorylation of phospholamban, a SERCA2a inhibitor, relieves SERCA inhibition, hence increasing sarcoplasmic reticulum Ca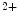 uptake. Downregulated expression of sarcolipin, another SERCA2a inhibitor, also offsets sarcoplasmic reticulum Ca
uptake. Downregulated expression of sarcolipin, another SERCA2a inhibitor, also offsets sarcoplasmic reticulum Ca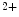 depletion caused by a Ca
depletion caused by a Ca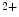 leak. Ryanodine receptor hyperphosphorylation or RYR gene mutations associated with catecholaminergic polymorphic ventricular tachycardia enhance RyR Ca
leak. Ryanodine receptor hyperphosphorylation or RYR gene mutations associated with catecholaminergic polymorphic ventricular tachycardia enhance RyR Ca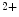 sensitivity [385].
sensitivity [385].
leak through RyR2s can result from: (1) excess sarcoplasmic reticulum Ca load reaching release threshold or (2) enhanced RyR Ca sensitivity. Hyperphosphorylation of phospholamban, a SERCA2a inhibitor, relieves SERCA inhibition, hence increasing sarcoplasmic reticulum Ca uptake. Downregulated expression of sarcolipin, another SERCA2a inhibitor, also offsets sarcoplasmic reticulum Ca depletion caused by a Ca leak. Ryanodine receptor hyperphosphorylation or RYR gene mutations associated with catecholaminergic polymorphic ventricular tachycardia enhance RyR Ca sensitivity [385].On the other hand, depolarization by 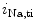 current can be antagonized by opposing transmembrane outward repolarizing currents (e.g.,
current can be antagonized by opposing transmembrane outward repolarizing currents (e.g., 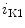 current through K
current through K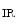 ; [385]). However, when the excitation threshold is reached, spontaneous action potentials arise.
; [385]). However, when the excitation threshold is reached, spontaneous action potentials arise.
< div class='tao-gold-member'>
current can be antagonized by opposing transmembrane outward repolarizing currents (e.g., current through K; [385]). However, when the excitation threshold is reached, spontaneous action potentials arise. Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


