Figure 20–1.
Potential mechanisms of fibrillation. Fibrillation may be initiated by a rapidly discharging, spontaneously active, ectopic focus (a), a single high frequency rotor (b) or by multiple functional reentrant circuits (c)
There is increasing awareness that arrhythmias do not usually begin in perfectly normal tissue, but require changes in tissue structure or function that constitute the arrhythmic substrate. The principal mechanisms underlying ectopic complex formation and reentry are illustrated in Fig. 20.2, along with the determinants of their substrates. Abnormal automaticity results from enhanced spontaneous phase 4 depolarization and causes premature beats that can trigger reentry in a vulnerable substrate. Early afterdepolarizations (EADs) are caused by failure of normal repolarization, prolonging the action potential (AP) plateau and allowing L-type Ca2+-channel reactivation to produce abnormal depolarization before repolarization can occur. Delayed afterdepolarizations (DADs) are due to spontaneous Ca2+-leak from sarcoplasmic reticulum (SR) Ca2+-stores. The released Ca2+ is exchanged for extracellular Na+ by the Na+, Ca2+-exchanger (NCX) in a fashion that generates a depolarizing current and a DAD. EADs and DADs can cause extrasystoles that trigger reentrant VF, but when repetitive can also cause sustained tachyarrhythmias that degenerate to VF. This chapter examines critically the substrates that underlie atrial (AF) and ventricular (VF) fibrillation, to inform the reader about their properties and to evaluate their similarities and differences.
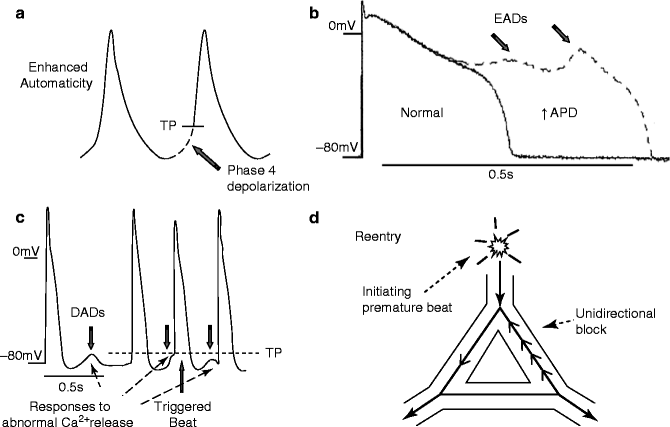
Figure 20–2.
Mechanisms of arrhythmia generation. (a) Enhanced automaticity: Abnormal impulse formation can result from a region of cardiac tissue prematurely brought to threshold potential (TP) by an increased slope of spontaneous phase 4 diastolic depolarization. (b) Early afterdepolarizations (EADs): Reductions in repolarizing currents and/or increases in depolarizing plateau currents can delay repolarization, prolonging action potential duration (APD). This can cause abnormal depolarizations (EADs, arrows) by reactivation of L-type Ca2+-channels. (c) Delayed afterdepolarizations (DADs): Under pathological conditions (eg, Ca2+ overload, ryanodine receptor dysfunction) Ca2+ is released from SR stores in diastole. This Ca2+ is exchanged by the Na+, Ca2+-exchanger for three times as many extracellular Na+ ions, producing an inward current. The inward current depolarizes the cell, producing delayed afterdepolarizations (DADs, downward arrows). If DADs are large enough to reach threshold potential (TP), they can trigger beats (upward arrow). (d) Reentry: Reentry may result from a premature initiating beat which fails to conduct in a region that is still refractory (unidirectional block), while conducting through an alternative connecting pathway that is no longer refractory. The impulse can then return backwards (retrogradely, dashed line) in the previously blocked pathway, and if enough time has elapsed for recovery of excitability, reenter through that pathway. Reentry requires a crucial balance between conduction and refractoriness to be maintained. It is favoured by brief refractory periods and slow conduction. Because variable refractoriness is needed to initiate reentry, spatial variability in refractoriness is an important predisposing condition
Biophysical Substrate
In biophysical terms, fibrillation represents self-sustaining rapid and irregular electrical activity. Fibrillation can be sustained by either a single rotor, a so-called “mother wave”, or by multiple smaller wavelets [4]. Mathematical modeling of AF shows that it can be sustained by either mechanism under different spatial distributions of acetylcholine [5], thus relating the mechanism to the degree of dispersion of refractoriness and regional ion channel expression. Ectopic complexes arising by automatic, DAD or EAD mechanisms contribute to the fibrillation substrate by providing the critical premature activation that initiates reentrant activity.
Figure 20.3a, b compare the features of the classical leading-circle reentry concept to those of the spiral-wave formalism, which is the presently-accepted biophysical representation of functional reentrant activity in cardiac tissue [6]. The transition between tachycardia and fibrillation proceeds through partial electrical wave break-up and the formation of secondary reentrant spiral waves that can either be sustained (consistent with the “multiple-wavelet hypothesis”, Fig. 20.1c) or transient (fibrillatory response to the “mother-wave”, Fig. 20.1b). An AP-based atrial model shows that either form of activity can underlie AF, depending on functional properties and tissue dimensions [7]. Figure 20.3c shows how a single primary rotor maintains fibrillation in the model. Figure 20.3d shows how, in a substrate with different properties, multiple unstable rotors can co-exist, each continuing for relatively brief periods of time (up to 1–2 s) but spawning sufficient daughter rotors before extinction to perpetuate AF. For further details and access to on-line movies, the interested reader is referred to the original paper [7]. Recently, it has been shown that generators have the tendency to be anchored to the regions of longer inexcitable period. Thus, the importance of geometry in AF maintenance was seen in situations of moderate anchoring for which rotor-core drifts between regions of longer inexcitable period exist, favoring generator disappearance at tissue borders [8].
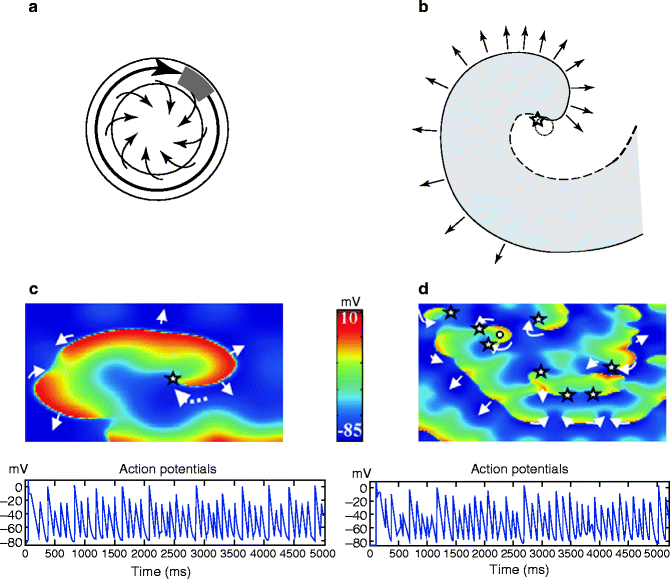
Figure 20–3.
(a) The leading circle model: Schematic diagram of a leading-circle reentry wave (shown here as a large black arrow). Activity establishes itself in the smallest pathway that can support reentry, given by the distance traveled by electrical activity in one refractory period (refractory period × conduction velocity). The length of this pathway is also called the wavelength for reentry. Inside the leading circle, centripetal wavelets (small arrows) emanating from the leading-circle wave front constantly maintain the central core in a refractory state. The wave tip and tail are separated by a region (shown in grey) that must be excitable, called the “excitable gap”. (b) Spiral-wave concept: Schematic diagram of a spiral wave with the activation front shown by a continuous line and the repolarization front by a dashed line. The point at which the continuous and dashed curves meet (shown with a star) has an undefined voltage-state, and is usually called the phase singularity point. There is no unexcitable core in the spiral wave model. (c) Example of fibrillation sustained by a single spiral wave. Top panel: Snapshot of transmembrane potential (colour coded: red fully depolarized tissue, blue resting tissue, yellow and green correspond to the AP plateau) during AF. The phase singularity of the single spiral is indicated by the star, and the dashed arrow shows the direction of rotation. The smaller arrows indicate the directions of electrical propagation. Bottom panel: Action potentials over 5 s, showing the non-periodic fibrillatory activity in a cell near the rotor core. (d) Example of fibrillation sustained by multiple spiral wave rotors. Top panel: Snapshot of transmembrane potential during AF. Generator rotors existing for between 100 ms and 1 s. are indicated by stars. In this case, longer-lasting rotors that maintained AF were always multiple and none of them lasted for long periods. However, they have generated daughter waves, some of which avoided annihilation and were able to maintain generator function when the initial generator extinguished. Bottom panel: Action potentials over 5 s, showing fibrillatory activity at the position pinpointed by the white circle in the top panel (For more detailed information and access to illustrative videos, see Zou et al. [7]) (Reproduced with permission of the American Physiological Society Inc.)
The first stage in the formation of a spiral wave is the occurrence of partial wave-block, equivalent to “unidirectional block” in classical re-entry theory. Wave-block occurs when the electrical wavefront encounters still-refractory tissue, creating a free wave-tip that re-enters through excitable tissue. Sustained spiral waves necessitate a sensitive balance between the time for the wavetip to re-enter and the refractory period, analogous to the conduction velocity-refractory period requirements for classical reentry. For a more detailed discussion of the relationship between classical reentry theory and the spiral-wave formulation, see Comtois et al. [9].
Two different substrate characteristics play a role in initiating and sustaining fibrillation. Refractoriness (static) heterogeneity can provide the conditions for re-entrant fibrillatory activity. Alternatively, dynamical spatial dispersion related to beat to beat variation in AP duration (APD) may be involved [10]. The APD restitution relation (relation between APD and recovery time) and conduction-velocity restitution relation are crucial determinants of this dynamical instability.
AF Substrate
Primarily non-dynamic mechanisms involving heterogeneity of ERP are thought to initiate breakup within atrial tissue [11]. Variations in neural innervation can also increase ERP heterogeneity. For example, remodeling of atrial electrical characteristics and innervation following left ventricular infarction, causing increased slope of the APD restitution curve, may promote AF via dynamical mechanisms [12]. Atrial shape details, including a range of complicated anatomical factors like pectinate muscles, pulmonary veins and limited inter-atrial connections, are likely important in AF, although the transmural electrical complexity of the ventricles is absent. Only as of recently are computational AF models being developed that consider the anatomical complexity of the atria [13].
The efficacy of Na+ channel blockade in terminating AF may be a unique feature of the atrial biophysical substrate. INa blockers terminate AF [14] via mechanisms demonstrated in AP-based computational models [15]. Na+-channel blockade slows AF and underlying rotors without increasing wavelength. Termination occurs because of increased size and meandering of the primary generator rotor, which becomes more likely to be annihilated against a boundary, as well as a dramatic decrease in the number of daughter wavelets that could provide new generators should the primary rotor extinguish [15, 16]. In contrast, INa blockers increase VF-related mortality in post-MI patients [17], suggesting a VF-promoting action. In ventricular computational models, qualitatively similar behaviours to those in AF are seen with VF, but spiral-wave stabilization occurs rather than termination [18]. Possible explanations of the failure of VF (in contrast to AF) to terminate with Na+-channel blockade are [1] spiral-wave stabilization by anchoring to structures like the papillary muscles [19] instead of terminating on boundary conditions, and [2] limited atrial-tissue mass that lacks the 3-dimensional transmural complexities of the ventricle and favours spiral-wave termination at boundaries.
VF Substrate
Wave breakup occurs in VF mainly via a dynamic mechanism, often first preceded by APD alternans, in particular discordant alternans. Although the importance of APD-restitution has been emphasized, recent work points to Ca2+-handling properties as the prime culprit [20, 21]. Tissue thickness and intramural reentry play a key role in VF [22]. Transmural heterogeneity and repolarization gradients [23] favour wave breakup and reentrant wave formation. Because of its tri-dimensionality, VF is based on scroll waves rotating around filaments, counterparts to spiral waves/core tips in AF. Mathematical modeling shows that transmural changes in ventricular fiber orientation (rotational anisotropy) can induce filament destabilization [24, 25] with filament-twisting leading to wave breakup [26]. Large twisting causes a transition to a turbulent regime characterized by a high density of moving filaments and secondary 3-dimensional spiral sources.
Summary of Biophysical Determinants
Similar general biophysical principles apply to AF and VF (Fig. 20.4). Important differences result primarily from discrepant structural characteristics of atria versus ventricles. The much greater thickness and transmural properties of the ventricles add an additional level of complexity to VF that may account for the discrepant responses to Na+-channel blockers, which terminate AF but increase the likelihood of VF.
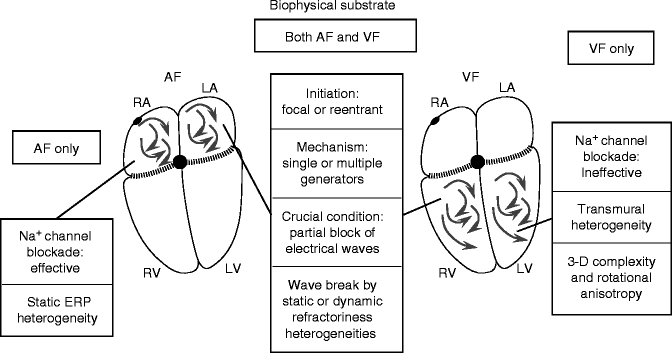
Figure 20–4.
A comparison of the biophysical properties of AF and VF substrates. The most important differences relate to the transmural complexities of the ventricle and the differential response to Na+-channel blockers
Ischemic Substrates
Ventricular tachyarrhythmias are the commonest cause of death, usually sudden, in the early phase of acute myocardial infarction (MI) [27]. Prior MI is a primary risk factor for VF [28]. AF also complicates acute MI quite regularly [29]. Acute ischemia promotes ion-channel dysfunction and tissue electrical dyssynchrony in relation to impairments in cell-to-cell coupling, intracellular acidosis, and accumulating extracellular K+. Coronary artery disease (CAD) is one of the conditions most commonly associated with AF [30]. The association could be due to secondary factors like congestive heart failure (CHF) caused by CAD, but could also be due to ischemia-induced AF.
AF Substrate
Atrial ischemia in itself promotes AF by impairing conduction, thus stabilizing atrial reentry that underlies AF and allowing AF to be much more readily sustained [31]. In contrast to ventricular MI [32], the effects of healed atrial MI have just been recently studied. Over time, tissue remodeling in chronic atrial ischemia creates substrates for both spontaneous ectopy, favouring reentry formation, and sustained reentry [33]. Border zone (BZ) tissue that separates ischemic from normal tissue, showed increased fibrous tissue content with augmentation in conduction heterogeneity. Studies of AF in small-animal MI models have described changes that more likely reflect the effects of post-MI hypertrophy and/or CHF than ischemia per se. Atrial fibrosis provides a substrate for AF in MI rats with CHF [34]. Chronic ventricular MI with no atrial involvement causes heterogeneous alteration of atrial electrical restitution related to atrial sympathetic hyperinnervation, a form of neural remodeling that provides a substrate for increased AF vulnerability [12].
VF Substrate
The presence of a zone of slow propagation and propagation block is an important common feature shared by the AF and VF substrates caused by acute myocardial ischemia/infarction. With subacute and healed ventricular infarction, a complex set of electrophysiological changes occur that create a substrate for ventricular tachycardia (VT) and VF [35]. Surviving cardiomyocytes in the MI border have reduced AP amplitude and phase 0 upstroke velocity (dV/dtmax) [36] and increased phase 4 depolarization slope, in addition to prolonged APD [37]. Most K+-currents are downregulated in post-MI border-zone cells [38]. These conditions are associated with enhanced automaticity and EADs [39].
Ca2+-handling is altered in border-zone and Purkinje cells post-MI, with ICaL diminished in several large animal models [40, 41], showing slower recovery [40] and hyperpolarizing shifts in inactivation voltage-dependence [41]. Cardiac contraction is based on Ca2+ released into the cytoplasm from SR Ca2+ stores in response to Ca2+ entry through L-type Ca2+ channels during the AP. The SR has specialized channels, called Ca2+-release channels or ryanodine receptors (RyRs, co-called because they were first isolated based on high affinity to the blocker ryanodine) that are responsible for releasing Ca2+ with the appropriate stimulus and then remaining closed until the next AP. Altered RyR function is importantly altered post-MI [42, 43], whereas NCX function appears unaltered [44]. These MI-induced disturbances in SR Ca2+-handling favour the occurrence of arrhythmogenic DADs.
Abnormalities of activation cause slow and often discontinuous conduction within and around the MI region [45, 46]. Marked changes in border-zone INa, including reduced current density, accelerated inactivation and slowed reactivation [47], impair conduction and excitability, promoting unidirectional block and reentry. Electrical conduction is also hindered by altered cell-to-cell coupling caused by gap junction dysfunction [46]. Gap junctions are smaller and less numerous in the peri-MI region [48]. Decreased side-to-side connections favour conduction block perpendicular to fiber orientation, producing a substrate for anisotropic reentry that underlies inducible VT/VF [49, 50]. Connexin proteins form the cell-to-cell connections in gap junctions that maintain low-resistance intercellular coupling. Altered expression of the principal ventricular connexin, connexin43, is a primary factor in post-MI gap-junction dysfunction and the substrate for VT/VF [49].
Summary of the Role of Myocardial Ischemia
Figure 20.5 summarizes the mechanisms demonstrated to provide substrates for AF and VF in relation to myocardial ischemia and infarction. A favourable AF and VF substrate is present at various phases of the acute ventricular ischemic process: immediately after acute MI, during the subacute phase and during the chronic healed phase. Important contributors include abnormalities in impulse propagation, heterogeneity in electrical properties, impairments in myocardial repolarization and disturbances in Ca2+-handling.
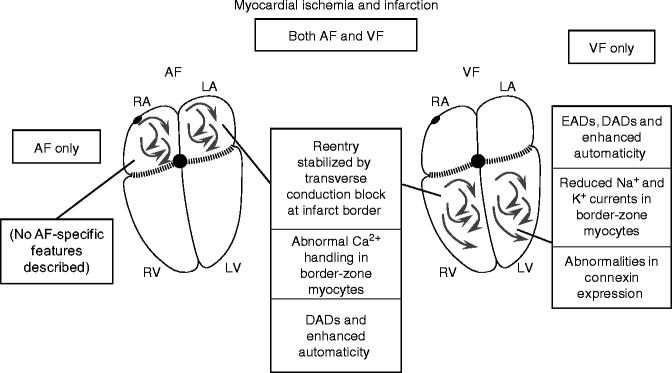
Figure 20–5.
Promotion of AF and VF by myocardial ischemia and infarction. Acute ischemia seems to affect the atria and ventricles similarly. There is nothing known about longer-term effects of previous ischemia/infarction on the atria. Vastly more work has been done on the ventricles
CHF
In the CHF population, sudden cardiac death, largely due to VF, is responsible for up to 50 % of CHF-associated mortality [51]. CHF is also one of the most common clinical causes of AF [52].
AF Substrate
Sinoatrial (SA) node function is abnormal in CHF [53, 54]. Bradycardia enhances refractoriness heterogeneity and thereby facilitates reentry [55], and can also promote the emergence of ectopic foci [56]. Atrial APD is unchanged or increased in CHF [57]. The only study available of atrial ionic-current changes in CHF showed decreased transient-outward current (Ito) and slow delayed-rectifier current (IKs), along with increased NCX current [57]. NCX carries a depolarizing diastolic current because it exchanges 1 intracellular Ca2+ ion (total charge +2) for 3 extracellular Na+ ions (total charge +3), carrying net inward current, when Ca2+ is extruded. This depolarizing current generates DADs, which can reach threshold and result in triggered activity, particularly in a setting of Ca2+-overload. There is evidence for DAD-induced focal atrial tachyarrhythmias in experimental CHF [58, 59]. These tachyarrhythmias may be very rapid and mimic AF. In addition, mapping studies have provided evidence for rapidly-discharging focal sources with fibrillatory conduction during AF in CHF dogs [60], as in Fig. 20.1a, suggesting that DAD-induced sustained triggered activity may be able to maintain AF.
Structural remodeling and fibrosis appear to be a significant factor in the CHF-induced AF substrate [61]. Localized regions of conduction slowing occur and manifest as increased conduction heterogeneity that favours AF [62, 63]. No significant overall atrial connexins Cx40 and Cx43 changes in mRNA and protein expression levels has been found but Cx43 dephosphorylation and redistribution toward transverse cell-boundaries has been documented [63]. In some cases, this has been shown to result in single macro-reentry circuits underlying AF [64], consistent with the mechanism shown in Fig. 20.1b. Recently, it has been proposed that heterogeneous spatial distribution of fibrosis at the posterior left atrial endocardium may govern AF dynamics driven by micro-reentry circuits [65].
VF Substrate
As in the atria, ventricular K+-currents, including both Ito and IKs, are down-regulated by CHF [66, 67]. Additionally, many studies show reduced IK1 [66]. One study showed reduced IKr [68], but most found IKr to be unchanged [66, 67]. Down-regulation of K+-currents increases APD (APD prolongation is a consistent feature in CHF [69]) and can lead to EAD-related tachyarrhythmias.
Significant Ca2+-handling changes occur in CHF ventricles. NCX expression and activity are both enhanced [70, 71]. CHF also causes SR Ca2+-leak due to abnormal RyR function [72]. Hyperphosphorylated RyRs, whether by protein-kinase A or Ca2+-calmodulin dependent protein-kinase II (CaMKII), are prone to abnormal spontaneous diastolic Ca2+-release [72]. CaMKII-induced RyR-hyperphosphorylation produces important SR diastolic Ca2+-leaks despite the decreased SR Ca load in CHF [72]. Ca2+ leaked from hyperphosphorylated RyRs is exchanged for extracellular Na+ by the NCX, producing DADs. These DADs can cause extrasystoles that may initiate reentry in a vulnerable VF substrate or directly lead to triggered-activity tachyarrhythmias that degenerate to VF [73]. In addition, spontaneous SR Ca2+ release promotes abnormal automaticity in latent pacemaker cells [74]. Ca2+-release induced DADs are enhanced by two other factors [70]: (1) increased NCX activity, which increases the amount of current generated for any level of Ca2+-release, and (2) downregulation of IK1, which increases membrane resistance, resulting in a larger depolarization for a given inward current.
Extensive ventricular structural remodeling occurs with end-stage CHF in humans [75] and experimental models [49]. Areas of interstitial fibrosis cause ventricular conduction slowing and promote reentry. However, CHF-related fibrosis is much more extensive in atria than ventricles [76], suggesting a more important role in AF than VF. Connexin43 expression is downregulated in human and experimental CHF [77–80]. Moreover, gap-junctional distribution and regulation are altered, with connexins redistributed to the cardiomyocyte lateral borders and dephosphorylated [78]. In animal models, along with tissue fibrosis, the changes in connexin43 expression and phosphorylation lead to conduction slowing [77, 78] and APD heterogeneity [80] in the failing heart, predisposing to reentry. In humans, HF resulted in the heterogeneous prolongation of APD, which significantly reduced the transmural APD gradients [81] along with heterogeneous remodeling of excitation-contraction coupling and calcium handling [82].
Summary of the Role of CHF
CHF creates a substrate for both AF and VF. Table 20.1 compares the underlying changes in the two tissues and Fig. 20.6 summarizes the known contributors to AF and VF substrates in CHF. Many similar alterations occur in each, and form substrates for EAD, DAD and reentry based arrhythmic activity. The main differences appear to be more prominent and functionally important fibrosis at the atrial level, and a clearer role for connexin alterations and EADs at the ventricular level.
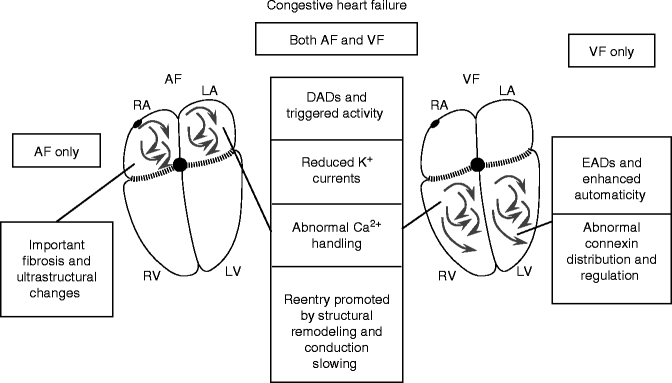
Table 20–1.
Substrate: congestive heart failure
Feature | AF | VF |
|---|---|---|
Potassium currents | ||
Ito | ↓ | ↓ |
IK1 | ↔ | ↓ |
IKr | ↔ | ↔ (↓) |
IKs | ↓ | ↓ |
Sodium currents | ||
INa | ? | ↓ |
Calcium handling | ||
ICaL | ↓ | ↓, ↔ |
NCX | ↑ | ↑ |
SERCA | ? | ↓ |
CaRC | ? | DS, Ph ↑ |
Structural changes | ||
Fibrosis | ↑↑↑ | ↑ |
Connexins | Ph↓, lateralization | ↓, lateralization |
APD | ↔ (↑) | ↑ |
CV | ↓ | ↓ |
EADs | ? | + |
DADs | + | + |
Reentry | + | + |
Figure 20–6.
CHF-related changes underlying AF and VF substrates. Many similar changes occur with CHF in both atria and ventricles. The roles of EADs and connexin changes are more clearly established in the ventricles
Genetic Factors
Recent advances in genetics have pinpointed a wide range of primary fibrillation-inducing mutations [83], which provide novel insights into molecular mechanisms. Two important fibrillation-promoting disorders of repolarization are the short QT syndromes (SQTSs) and long QT syndromes (LQTSs). SQTS causes early-onset paroxysmal AF [84] and VF [85]. The prolonged APD in LQTS patients often leads to the EAD-associated polymorphic ventricular tachyarrhythmia Torsades de Pointes, TdP, which can degenerate to VF [86]. Arrhythmic mutations can also affect conduction properties, like the loss-of-function mutations in cardiac Na+-channels that cause the arrhythmic Brugada Syndrome [87] and probably also AF [88]. Inherited arrhythmia predilections can also affect Ca2+-handling and connexin function.
AF Substrate
While SQTS most strikingly causes VF-related sudden death, SQTS clearly also predisposes to AF [84, 85, 89, 90]. The AF substrate is presumably based on accelerated repolarization, which makes the atria highly vulnerable to reentry, as seen with many acquired arrhythmia paradigms [91]. Atrial and ventricular ERPs are very short and both AF and VF are readily inducible in SQTS [84]. A KCNQ1 mutation causes familial AF by increasing IKs and giving it a time-independent behaviour [92]. A mutation in KCNE2, a β-subunit of uncertain function, has been described in familial AF kindred [93]. Mutated subunits produce gain-of-function in a time-independent IKs component upon co-expression with KCNQ1. It is not clear why these two gain-of-function IKs mutations do not cause SQTS and ventricular tachyarrhythmias. Loss-of-function mutations in genes encoding the cardiac L-type calcium channel (CACNA1C and CACNB2b) have also been associated with a familial sudden cardiac death syndrome and increase AF sensitivity [94]. There is some evidence suggesting that LQTS can lead to AF [95], but this remains controversial. Recently, other paradigms of AF apparently associated with delayed repolarization have been described [96, 97]. Prolonged atrial APs could lead to AF either by EAD-mediated mechanisms in susceptible patients or by favouring wave-front breakup at critical rates [96].
Slow conduction favours reentry by leaving more time for recovery of excitability in potential reentrant pathways (Fig. 20.2). Conduction can be slowed by altering the Na+-current that provides the energy for electrical conduction or by affecting cell-to-cell coupling. Several mutations in SCN5A, which encodes the cardiac Na+-channel, lead to a variety of phenotypes including AF [88]. A single-nucleotide polymorphism (SNP) in the connexin40 promoter that reduces Cx40 transcription is associated with AF vulnerability [98, 99]. Gollob et al. have recently reported the intriguing observation that several patients with idiopathic, early-onset AF have somatic mutations in Cx40 [100].
VF Substrate
SQTS produces transmurally-heterogeneous APD abbreviation by preferentially abbreviating repolarization in M-cells, promoting reentry [101]. A variety of loss-of-function K+ channel mutations, or gain-of-function Na+ and Ca2+ channel mutations can lead to impaired repolarization, LQTS, TdP and VF precipitation [102].
The Brugada Syndrome is characterized by ST segment elevation in right precordial leads, right bundle branch block, and susceptibility to VF [87]. Loss-of-function SCN5A genes are causal in ∼25 % of Brugada syndrome patients [103]. Loss of SCN5A function can promote reentry by slowing conduction. However, a major component of the pathophysiology in the Brugada Syndrome appears to be loss of the action potential plateau in the epicardium, where large Ito can cause very early repolarization in the absence of countervailing INa, with current spread from normal AP plateaus in the endocardium causing “phase 2 reentry” (for detailed review, see Antzelevitch et al. [104]).
A variety of genetic syndromes promote VF by impairing Ca2+-handling. Catecholaminergic polymorphic ventricular tachycardia (CPVT) mutations affect Ca2+-handling and buffering. Missense mutations in the CASQ2 gene, encoding the principal SR Ca2+-buffer calsequestrin, are associated with autosomal-recessive CPVT [105]. Mutations in the gene encoding RyRs are the most common cause of CPVT [106]. Alterations in these CPVT-related gene products cause abnormal diastolic SR Ca2+ release, producing DADs. Catecholamine-induced β-adrenergic receptor stimulation promotes VT/VF in CPVT patients by increasing Ca2+-entry through L-type Ca2+-channels, increasing Ca2+-loading and enhancing spontaneous diastolic Ca2+-release and DADs. Ankyrin-B mutations disrupt the cellular localization of a variety of proteins, including Na+/K+-ATPase and NCX, and ankyrin-B deficiency produces defects in Ca2+ handling and afterdepolarizations that may be responsible for fibrillation-induced cardiac death, as well as AF [107, 108].
Summary of the Role of Genetic Determinants
There is considerable overlap in genetic AF and VF determinants (Fig. 20.7), but the unique role of connexin40 in the atrium is reflected in the lack of ventricular arrhythmias with connexin40 gene abnormalities. Repolarization abnormalities predispose much more clearly to VF than AF, presumably because of the much longer AP durations in Purkinje and M cells than the atrium, predisposing to EADs and repolarization heterogeneity. CPVT-related mutations also seem to be much more likely to lead to VF than AF.
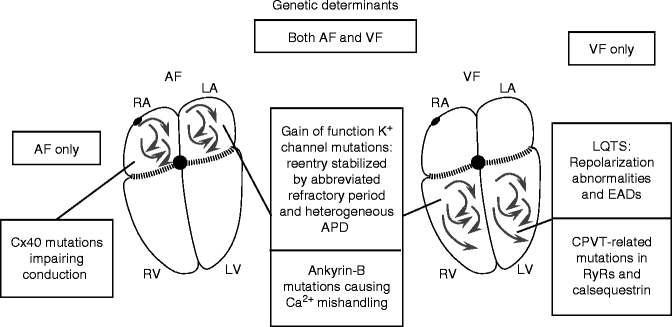
Figure 20–7.
Genetically determined AF and VF substrates. The role of connexin40 abnormalities in AF is clear, as is the role of SQTS, LQTS and CPVT mutations in VF. In addition, gain-of-function K+-channel mutations have been described that appear to cause AF but not VF, for presently unclear reasons. Ankyrin-B mutations cause both AF and VF. Recently, repolarization-delaying genetic alterations have been described as associated with AF, but the precise mechanisms remain unclear
Neuroregulatory Factors
The autonomic nervous system (ANS) has long been recognized to play a potentially important role in AF and VF [109, 110]. The spatial heterogeneity of ANS actions increases refractoriness dispersion [111], favouring fibrillation. This is particularly important in vagal AF, a well-recognized clinical entity [112]. Alterations in neurohormonal function can also play a critical modulating role for fibrillation substrates. For example, adrenergic stimulation is a well-recognized precipitator of ventricular tachyarrhythmias in LQTS and CPVT. β-Adrenergic stimulation is also an important contributor to ischemic VF, since β-adrenoceptor blockers are among the only drugs known to prevent VF post-MI in patients with coronary artery disease [113].
AF Substrate
Vagal stimulation produces high vulnerability to sustained AF [114]. Spatially-heterogeneous refractoriness abbreviation appears particularly important in the pathogenesis of vagal AF [115]. The local (intrinsic) cardiac nervous system is affected by pathology associated with AF [116]. Atrial arrhythmias can be induced by stimulating mediastinal nerve branches of the thoracic vagosympathetic complex [117], or mediastinal nerves associated with pulmonary veins [118, 119].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


