MicroRNA
Tumorigenesis
IPF development
Related molecules
miR-21
↑
↑
TPM1, PDCD4, PTEN,TGFβ, NFIB, mapsin, Spry-2, MARCKS
MMPs, RECK, SMAD2, SMAD7, FGF2
Let-7
↓
↓
TGFβ, HMGA2, SMAD3, RAS, CDC25A
miR-155
↑
↑
TGFβ, KGF7, SMARCA4, TP53INP1, PTEN, PDCD4, SHIP1
miR-29
↓
↓
TGFβ, SMAD3, PI3K/AKT, DNMT3A, DNMT3B
miR-30
↓
↓
RAS, Rab18, TGFβ, WISP1
miR-210
↑?
↑
Hypoxia, MNT, FGFRL1
miR-199a–5p
↑
↑
TGFβ, CAV1, SMARCA4
miR-145
↓
↑
EGFR, NUDT1, TGFβ, KLF4
miR-200
↓
↓
ZEB1, ZEB2, Sec23a, IL-8, CXCL1, TGFβ, GATA3, PTEN
BIM, TGFβRII, CTGF, RB, and p21
miR-17–92
↑
↓
DNMT-1, cMYC, E2F1/E2F3, STAT3
miR-375
↓
↓
TGFβ/SMAD, CLDN1, FZD8, Wnt/β catenin
miR-185
↓
↑?
CDK6, AKT1, AGO1, AGO2, RISCs
miR-154
↓?
↑?
NFκB, HIP-1, MAPK, Notch, TGFβ, p15, DKK2, DIXDC1
PPP2CA, FZD 4/5/6, LRP, KREMEN1, Wnt/β catenin
14.6 Signaling Pathways
IPF is characterized by injured and hyperplastic alveolar epithelium, which releases diverse molecules including growth factors, cytokines, and MMPs, causing the activation and proliferation of mesenchymal cells, ECM deposition, and the accumulation of fibroblasts. These processes can lead to basal membrane disruption, fibrin formation, abnormal wound repair, and angiogenesis through multiple signaling pathways, thereby promoting cellular apoptosis or migration. In many lung cancers, these signals can also activate oncogenes or inactivate tumor suppressor genes, leading to oncogenesis through aberrant oncogenic pathways. These findings suggest that common signaling pathways may contribute to the development of both IPF and lung cancer. Indeed, some individual key molecules, as described in the previous sections, are associated with both oncogenesis and fibrogenesis through common signaling pathways (Fig. 14.1). Therefore, we further describe noteworthy signaling pathways mentioned in the previous sections.
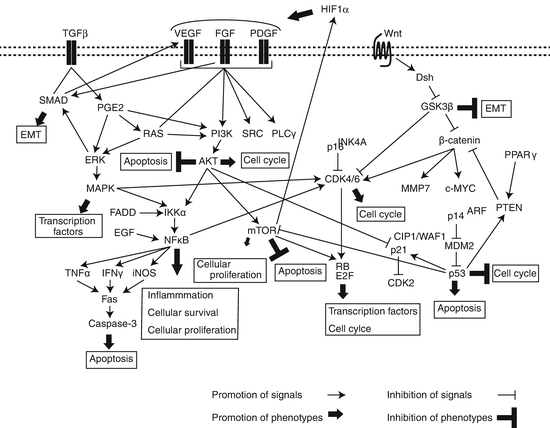
Fig. 14.1
Potential signaling pathways underlying the common pathogenesis of IPF and lung cancer
14.6.1 Transforming Growth Factor-β Signaling
As described in the previous sections, the TGFβ signaling pathway plays important roles in wound healing and fibrogenesis through its cross talk with other signaling pathways and diverse mediators. TGFβ-induced EMT and accumulation of myofibroblasts can be promoted by integrin-dependent activation of SMAD and Wnt/β-catenin signaling [103]. Myofibroblasts are resistant to apoptosis, which promotes fibrogenesis, partly through reduction of the anti-fibrotic prostaglandin E2 (PGE2) in IPF [104]. The features of these phenomena that lead to EMT appear to be similar to those of tumor cells, and myofibroblasts promote cellular infiltration and progression through multiple mediators in a tumor environment.
14.6.2 Wnt Signaling
The Wingless-type protein (Wnt) family, which consists of 19 secreted proteins, transmits its signal through activation of multiple pathways. One major Wnt pathway, termed the canonical pathway, inhibits GSK3β, leading to the stabilization and accumulation of β-catenin in the cytosol and its translocation into the nucleus, consequently resulting in enhanced expression of target genes that are associated with diverse diseases. As described in the previous Sect. 14.3.1, the Wnt/β-catenin pathway is one target of TERT regulation. β-catenin transmission of Wnt signals to the nucleus regulates diverse oncogenic genes including cyclin D1 and cMYC, and its overexpression and stabilization can promote oncogenesis. The review by Stewart et al. indicates that expression of WNT1, a Wnt ligand, is correlated with aberrant β-catenin expression and increased expression of cMYC, cyclin D1, VEGFA, MMP7, Ki67, and survivin, resulting in proliferation of NSCLC [105]. The other Wnt pathway, termed the noncanonical pathway, which does not involve β-catenin, is mainly comprised of two types of pathways, the planar cell polarity pathway (Wnt-PCP pathway) and the Wnt-calcium pathway (Wnt/Ca2+ pathway). The combined expression of WNT7A and FZD9 in NSCLC cell lines leads to ERK5 activation, which in turn leads to increased PPARγ expression and activation of Sprouty-4. Sprouty-4 inhibits EMT, consequently inhibiting tumor growth [106, 107]. Indeed, aberrant expression of Wnt ligands and Wnt signaling mediators has been frequently found in NSCLC.
The Wnt/β-catenin pathway also promotes EMT and myofibroblast differentiation that is mediated through TGFβ, thereby contributing to pulmonary fibrogenesis. The Wnt/β-catenin signaling pathway was upregulated in lung tissues of IPF patients. Moreover, β-catenin targets cyclin D1 and MMP7, both of which are considered to be associated with pulmonary fibrogenesis [108].
14.6.3 PI3K/AKT/mTOR Signaling
Phosphoinositide 3-kinases (PI3Ks) regulate cellular proliferation, survival, adhesion, and motility. These kinases stimulate PIP3 synthesis on the cell membrane, thereby promoting the PI3K/AKT/mTOR pathway, a downstream signaling pathway composed of multiple receptor tyrosine kinases. The PI3K/AKT/mTOR pathway is activated early in the pathogenesis of pulmonary tumorigenesis through the modulation of multiple signals as follows, resulting in inhibition of apoptosis and cell survival: (1) mutations in PI3K or PTEN as well as in the EGFR or KRAS and (2) PIK3CA amplification, PTEN loss, or AKT activation [109]. In fibroblasts of IPF, PI3K/AKT/mTOR signals are aberrantly activated, while PTEN activity is lower than that of normal cells [110]. Furthermore, the PI3K/AKT/mTOR pathway interacts with many other signaling pathways, including those of MAPK and VEGFR, in IPF cells [111].
14.6.4 Nuclear Factor-κB Signaling
Cross talk between the NFκB and PI3K/AKT/mTOR signaling pathways may contribute to lung cancer cell survival and proliferation [112]. PI3K/AKT promotes NFκB activity in an IKKα-dependent manner [113], while NFκB is activated by Fas-associated death domain protein (FADD) phosphorylation through IKK, thereby promoting proliferation in lung adenocarcinoma [114]. Furthermore, EGF induces IKK-independent NFκB activation through IκBα phosphorylation in lung adenocarcinoma cells [115]. NFκB activation was associated with KRAS mutation, and both lost p53 function and active KRAS G12D , the major RAS point mutation in lung cancer, constitutively activate NFκB as well as downstream signaling pathways, such as PI3K and MAPK, in lung cancer cells [116]. NFκB activation is also found in the IPF-associated inflammatory process. NFκB-dependent inflammatory mediators, including tumor necrosis factor (TNF)-α, TGFβ, interleukin (IL)-1, inducible nitric oxide synthase (iNOS), and interferon (IFN)-γ, were highly expressed in IPF patients and in animal models of pulmonary fibrosis [117–119]. Previous in vivo studies showed that suppression of the NFκB signaling pathway can attenuate bleomycin-induced pulmonary fibrosis, while in vitro studies also found that the NFκB signaling pathway contributed to the regulation of TGFβ [119–121].
14.6.5 Fibroblast Growth Factor Signaling
Fibroblast growth factors (FGFs) are a family of homologous polypeptide ligands that bind to their cognate FGF receptors (FGFRs) [122]. There are 22 FGF ligands comprising six subfamilies and four FGFR isoforms that are differentially activated by FGF ligands in conjunction with the scaffold protein heparan sulfate proteoglycan. Binding of FGF ligands to FGFRs transmits signals associated with angiogenesis as well as with cellular migration and proliferation, and the FGF signaling pathway is involved in the pathogenesis of diverse diseases. However, FGF signaling exerts reciprocal effects on tumor promoting and suppressive activities, which are dependent on the tumor type. Regarding IPF, TGFβ released from AECs induces the proliferation of pulmonary fibroblasts through FGF2, which promotes phosphorylation of p38 MAPK and JNK [123], and high FGF2 levels have been found in the bronchoalveolar lavage fluid and serum of IPF patients [124]. Many NSCLC cells showed elevated FGF2 levels, which in turn promote the growth of these tumor cells by intracrine mechanisms [125]. FGF2 also contributes to angiogenesis and tumor proliferation in many types of cancer, and FGF2-mediated autocrine and paracrine signaling can potently promote oncogenesis.
Another FGF, FGF1, exerts antifibrotic activity through downregulation of collagen expression and antagonization of some TGFβ-mediated profibrotic functions [126]. FGF1 was also shown to restore TGFβ-induced EMT in AECs through dephosphorylation of the SMAD2 phosphorylation that was induced by the MEK/ERK pathway. FGF1-mediated activation of FGFR signaling induces the recruitment and activation of SRC homology (SH2)- or phosphotyrosine (PTB)-containing proteins, thereby activating multiple signaling pathways including PI3K/AKT, MEK1/2-ERK, and p38MAPK [127]. Recently, Daly et al. reported that low levels of FGF1 are associated with favorable outcomes in stage I lung adenocarcinoma [128].
14.6.6 Fas/FasL Signaling
Fas, a cell surface death receptor of the TNF receptor (TNFR) superfamily, paradoxically promotes both survival/differentiation and apoptosis through the modulation of immune responses. Low to absent staining of Fas was detected in fibroblastic cells of fibroblast foci from IPF patients [129]. Exposure to the proinflammatory cytokines, TNF-α and IFNγ, increases Fas expression in an NFκB- and MAPK-dependent manner, thereby sensitizing fibroblasts to Fas-induced apoptosis and reversing the resistance of lung fibroblasts to apoptosis, which is mediated by a prosurvival effect of TGFβ. Aberrant Fas/Fas ligand (FasL) expression is found in many lung cancer cells and samples [130], suggesting its contribution to lung carcinogenesis.
14.6.7 Vascular Endothelial Growth Factor Signaling
The vascular endothelial growth factor (VEGF) signaling pathway is well characterized. VEGF ligands (VEGFA to VEGFE and placental growth factor) induced by hypoxia, growth factors, and cytokines interact with VEGF receptors (VEGFR1–3), especially with VEGFR2 that activates the PI3K/AKT pathway; promotes cellular proliferation, migration, and survival as well as angiogenesis; and inhibits apoptosis. VEGF is highly expressed in lung cancer, and its expression is associated with poor prognosis [131]. While normal wound healing requires angiogenesis, aberrant neovascularization is often found in IPF tissues, suggesting that VEGF signaling is associated with the development of IPF. Furthermore, Kobayashi et al. showed that SMAD3 signaling mediates TGFβ-induced VEGFA production in human lung fibroblasts [132].
14.6.8 Platelet-Derived Growth Factor Signaling
The platelet-derived growth factor (PDGF) family, which consists of PDGFA to PDGFD, promotes cellular proliferation, invasion, and angiogenesis as well as wound healing and proliferation of mesenchymal cells. Enhanced PDGF expression and activation of its signaling have been reported in different types of cancer and in fibrotic disorders. Dimeric PDGFs bind to the receptors PDGFRα and PDGFRβ, thereby transmitting signaling through PI3K, SRC, phospholipase C-γ, and RAS pathways in both an autocrine and a paracrine manner [133]. PDGFA and PDGFC paracrine signaling was associated with fibroblastic tumor infiltration in NSCLC cell lines [134]. Furthermore, PDGF signaling, which partly overlaps with VEGF signaling, activates cancer-associated fibroblasts (CAFs) and recruits VEGF-producing stromal fibroblasts, leading to both tumorigenesis and angiogenesis [135]. PDGF that is synthesized by alveolar macrophages is a potent mitogenic and chemotactic factor for fibroblasts. Previous in vitro studies showed that PDGF interacts with several fibrotic mediators including TGFβ, IL-1, TNF-α, FGF, and thrombin, thereby promoting fibrogenesis. Macrophages and fibroblasts from IPF patients produce PDGF, whereas PDGFB and PDGFR mRNAs are highly expressed in hyperplasic ATII cells in IPF [136]. PDGF/PDGFR signaling can at least partly interact with VEGF/VEGFR and FGF/FGFR signaling pathways, and the intricate cross talk between these pathways may contribute to the common pathogenesis of IPF and lung cancer.
14.6.9 RAS/RAF/MEK/MAPK Signaling
Signaling of the RAS/RAF/MEK/MAPK pathway is one of the most extensively characterized signals and modulates cellular migration, proliferation, and apoptosis; its aberrant signals are therefore involved in the development of diverse diseases. In particular, this pathway is frequently activated in lung cancer, most commonly via KRAS mutations in approximately 20 % of lung cancers, particularly in adenocarcinomas in smokers [137]. The RAS/RAF/MEK/MAPK pathway has also been associated with the development of IPF. Many molecules and pathways, as well as most signals described in this section, can converge, regulate, or interact with the RAS/RAF/MEK/MAPK pathway.
14.6.10 Signaling Through Connexin
Gap junctions are composed of protein complexes including connexins that characterize cell-cell communication, and their alteration is tightly associated with cellular proliferation, tissue repair, and tumor growth. Vancheri et al. reported the possible role of connexins, especially connexin 43 (CX43), in the common pathogenesis of IPF and lung cancer [138].
Multiple pathways that are mutually dependent interact with one another, thereby regulating the signals that can lead to inflammatory processes and tumorigenesis. Therefore, a possible common pathway of IPF and lung cancer may provide a therapeutic perspective on these comorbid disorders. For example, nintedanib that targets PDGFRα and PDGFRβ, VEGFR1 to VEGFR3, and FGFR1 to FGFR3 has therapeutic potential for both IPF and NSCLC based on the phase III trials, INPULSIS-1 and INPULSIS-2, for IPF and LUME-Lung 1 for NSCLC [139, 140].
14.7 Conclusion
Both IPF and lung cancer are heterogeneous disease groups, and furthermore, multiple pathways and diverse molecules that interact with one another can be associated with both diseases, raising the complexity of the pathogenesis of these diseases. However, we often encounter patients with lung cancer comorbid with IPF in clinical practice, and as described in Sect. 14.1, lung cancer occurs in 4.4–38 % of patients with interstitial pneumonia according to previous reports. Possible commonality in the pathogenesis of IPF and lung cancer has been discussed in this chapter, suggesting that in this context, there may be a common pathway that contributes to the development of IPF and lung cancer. Diverse signals and molecules can transmit individual signaling pathways, converge on a common pathway, and promote inflammatory processes in IPF and lung tumorigenesis. These phenomena may lead to a high frequency of comorbid IPF and lung cancer. Therefore, an efficacious therapeutic strategy for both diseases that targets such a possible common pathway may be exploited in the future, although it has already been attempted, in part. In particular, although the incidence of lung cancer with IPF is steadily increasing, there are few reports regarding its treatment. For more efficacious and efficient therapy for IPF and lung cancer, a more fundamental pathway that underlies the development of both diseases needs to be determined.
References
1.
Kawasaki H, Nagai K, Yokose T, Yoshida J, Nishimura M, Takahashi K, et al. Clinicopathological characteristics of surgically resected lung cancer associated with idiopathic pulmonary fibrosis. J Surg Oncol. 2001;76(1):53–7.PubMed
2.
Lawson WE, Loyd JE. The genetic approach in pulmonary fibrosis: can it provide clues to this complex disease? Proc Am Thorac Soc. 2006;3(4):345–9.PubMedCentralPubMed
3.
Armanios MY, Chen JJ, Cogan JD, Alder JK, Ingersoll RG, Markin C, et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med. 2007;356(13):1317–26.PubMed
5.
Jang JS, Choi YY, Lee WK, Choi JE, Cha SI, Kim YJ, et al. Telomere length and the risk of lung cancer. Cancer Sci. 2008;99(7):1385–9.PubMed
6.
McKay JD, Hung RJ, Gaborieau V, Boffetta P, Chabrier A, Byrnes G, et al. Lung cancer susceptibility locus at 5p15.33. Nat Genet. 2008;40(12):1404–6.PubMedCentralPubMed
7.
Ma X, Gong R, Wang R, Pan Y, Cai D, Pan B, et al. Recurrent TERT promoter mutations in non-small cell lung cancers. Lung Cancer. 2014;86(3):369–73.PubMed
8.
Liu T, Hu B, Chung MJ, Ullenbruch M, Jin H, Phan SH. Telomerase regulation of myofibroblast differentiation. Am J Respir Cell Mol Biol. 2006;34(5):625–33.PubMedCentralPubMed
9.
Wang Y, Kuan PJ, Xing C, Cronkhite JT, Torres F, Rosenblatt RL, et al. Genetic defects in surfactant protein A2 are associated with pulmonary fibrosis and lung cancer. Am J Hum Genet. 2009;84(1):52–9.PubMedCentralPubMed
10.
Nogee LM, Dunbar 3rd AE, Wert SE, Askin F, Hamvas A, Whitsett JA. A mutation in the surfactant protein C gene associated with familial interstitial lung disease. N Engl J Med. 2001;344(8):573–9.PubMed
11.
Jiang F, Yin Z, Caraway NP, Li R, Katz RL. Genomic profiles in stage I primary non small cell lung cancer using comparative genomic hybridization analysis of cDNA microarrays. Neoplasia. 2004;6(5):623–35.PubMedCentralPubMed
12.
Rodenhuis S, Slebos RJ. Clinical significance of ras oncogene activation in human lung cancer. Cancer Res. 1992;52(9 Suppl):2665s–9.PubMed
13.
Takahashi T, Munakata M, Ohtsuka Y, Nisihara H, Nasuhara Y, Kamachi-Satoh A, et al. Expression and alteration of ras and p53 proteins in patients with lung carcinoma accompanied by idiopathic pulmonary fibrosis. Cancer. 2002;95(3):624–33.PubMed
14.
Verri C, Roz L, Conte D, Liloglou T, Livio A, Vesin A, et al. Fragile histidine triad gene inactivation in lung cancer: the European Early Lung Cancer project. Am J Respir Crit Care Med. 2009;179(5):396–401.PubMed
15.
Rabinovich EI, Kapetanaki MG, Steinfeld I, Gibson KF, Pandit KV, Yu G, et al. Global methylation patterns in idiopathic pulmonary fibrosis. PLoS One. 2012;7(4):e33770.PubMedCentralPubMed
16.
Cooper WA, Lam DC, O’Toole SA, Minna JD. Molecular biology of lung cancer. J Thorac Dis. 2013;5 Suppl 5:S479–90.PubMedCentralPubMed
17.
White ES, Atrasz RG, Hu B, Phan SH, Stambolic V, Mak TW, et al. Negative regulation of myofibroblast differentiation by PTEN (Phosphatase and Tensin Homolog Deleted on chromosome 10). Am J Respir Crit Care Med. 2006;173(1):112–21.PubMedCentralPubMed
18.
Wang XM, Zhang Y, Kim HP, Zhou Z, Feghali-Bostwick CA, Liu F, et al. Caveolin-1: a critical regulator of lung fibrosis in idiopathic pulmonary fibrosis. J Exp Med. 2006;203(13):2895–906.PubMedCentralPubMed
19.
Racine C, Belanger M, Hirabayashi H, Boucher M, Chakir J, Couet J. Reduction of caveolin 1 gene expression in lung carcinoma cell lines. Biochem Biophys Res Commun. 1999;255(3):580–6.PubMed
20.
Engelman JA, Wykoff CC, Yasuhara S, Song KS, Okamoto T, Lisanti MP. Recombinant expression of caveolin-1 in oncogenically transformed cells abrogates anchorage-independent growth. J Biol Chem. 1997;272(26):16374–81.PubMed
21.
Ho CC, Huang PH, Huang HY, Chen YH, Yang PC, Hsu SM. Up-regulated caveolin-1 accentuates the metastasis capability of lung adenocarcinoma by inducing filopodia formation. Am J Pathol. 2002;161(5):1647–56.PubMedCentralPubMed
22.
Yoo SH, Park YS, Kim HR, Sung SW, Kim JH, Shim YS, et al. Expression of caveolin-1 is associated with poor prognosis of patients with squamous cell carcinoma of the lung. Lung Cancer. 2003;42(2):195–202.PubMed
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


