Fig. 16.1
Timeline of drugs approved for treatment of pulmonary arterial hypertension in the USA. PGI 2 prostaglandins, ERAs endothelial receptor antagonists, PDE5Is phosphodiesterase type-5 inhibitors, sGC+ soluble guanylate cyclase stimulator
Medications Presently Available for Treatment of PAH
There are currently four classes of medications approved for the treatment of PAH. The first of these are the prostacyclin analogues. Prostacyclin (PGI2) was discovered by Sir John Vane in the late 1970s [4] and is a member of the prostaglandin family of peptides and is synthesized in platelets and vascular endothelial cells. It has potent vasodilatory effects on vascular smooth muscle and anti-aggregation effects on platelets. The expression of prostacyclin synthase, the major enzyme involved in the synthesis of prostacyclins, is decreased in the lungs of PAH patients as is the ratio of circulating PGI2 to thromboxane, suggesting a state of relative PGI2 deficiency [5, 6]. Intravenous prostacyclin was found to be a potent pulmonary vasodilator and its ability to improve functional capacity and pulmonary hemodynamics in patients with PAH led to its development as pharmaceutical agent. In 1995, the synthetic PGI2, epoprostenol, became the first FDA-approved treatment for PAH. The need for continuous intravenous infusion via central venous access makes epoprostenol a challenging therapy for most patients and its risk of catheter-related infection has led to the development of alternative prostacyclin therapies for PAH. Treprostinil is a prostacyclin derivative that is stable at room temperature and has a considerably longer half-life than epoprostenol. It was designed for continuous subcutaneous infusion, thereby obviating the need for a central venous catheter. This approach was found to be effective, but is associated with significant pain at the site of infusion making it difficult for some patients to continue treatment. Since its initial release in 2001, treprostinil has been approved for intravenous, inhaled, and oral administration. Iloprost is another prostacyclin derivative that is approved in the USA as an inhalational therapy for treating PAH, although its shorter half-life compared to treprostinil has resulted in the need for more frequent treatments. All three prostanoids have been shown to be efficacious at improving pulmonary hemodynamics, 6MWD, and/or WHO functional class in PAH, although the efficacy data for inhaled and oral prostacyclin therapy do not appear to be as robust as for intravenous or subcutaneous infusion [7–10].
Increased expression of endothelin-1 (ET-1), a potent vasoconstrictor and smooth muscle mitogen, has also been implicated in the pathogenesis of PAH. ET-1 is expressed at high levels in the pulmonary vascular lesions of PAH patients [11]. Plasma ET-1 levels are increased in patients with PAH compared to controls and correlate with the severity of PH [12, 13]. The biologic activity of ET-1 is mediated by endothelin receptor A and endothelin receptor B (ETA and ETB). Both nonselective endothelin receptor antagonists (ERAs) and antagonists selective for ETA have been developed for the treatment of PAH and have been shown to improve pulmonary hemodynamics and 6MWD and to delay time to clinical worsening [14, 15].
The secondary messenger cGMP plays important roles in modulating pulmonary hypertensive and right ventricular hypertrophic responses. Intracellular cGMP levels are increased by activation of soluble and particulate guanylate cyclases in response to nitric oxide (NO) and the natriuretic peptides, respectively. A considerable body of evidence has accumulated to suggest that decreased bioavailability of NO contributes to the pathogenesis of PAH (for review, see Klinger et al. [16]). Circulating levels of atrial and brain natriuretic peptide (ANP and BNP) are increased in patients with PAH, but the ratio of urinary cyclic GMP/BNP is markedly decreased suggesting downregulation of the particulate guanylate cyclase, natriuretic peptide receptor-A [17]. cGMP levels are also regulated by the activity of a family of phosphodiesterases (PDE) that are primarily responsible for its degradation. PDE type-5 (PDE5) is the primary enzyme responsible for degradation of cyclic GMP in the lung. Its activity has been shown to be increased in animal models of PH [18]. Drugs that inhibit PDE5 such as sildenafil and tadalafil have been shown to be effective at lowering PVR and improving 6MWD in patients with PAH [19, 20] and have been approved for treatment. Other drugs, such as inhaled NO or the recently approved soluble guanylate cyclase stimulator, riociguat that stimulate the synthesis of cGMP, have also been found to be effective treatments for PAH [21].
Rationale for Combination Therapy
There are two major reasons that are most often cited for combining different therapies in the treatment of PAH. The first is that numerous mechanisms contribute to the pathogenesis of PAH and therefore multiple signaling pathways need to be targeted simultaneously. The second is that there are potential interactions between the known classes of PAH-specific medications that could provide additive or synergistic effects when used together. For example, many of the beneficial effects of prostacyclins on the pulmonary circulation are mediated by intracellular cAMP levels. Cyclic GMP acts as a substrate for some of the same phosphodiesterases that metabolize cyclic AMP and elevation of intracellular cGMP by PDE-5 inhibitors may slow the metabolism of cAMP. Indeed, studies have shown that PDE inhibitors that increase intracellular levels of cGMP in the RV of rats with monocrotaline-induced pulmonary hypertension also result in an increase in cAMP levels that increase right ventricular contractility [22]. Thus, combining prostacyclin therapy with a PDE5 inhibitor may result in better elevation of both cyclic nucleotide levels and potentially increase their clinical efficacy. Similarly, NO and the natriuretic peptides have been shown to suppress synthesis of endothelin, likely via elevation of intracellular cyclic GMP levels [23, 24]. Thus, a combination of a PDE5 inhibitor and an ERA may be more effective at blunting the effects of endothelin than either agent alone.
Clinical Trials of Combination Therapy
The pharmacologic approach to treating PAH has evolved considerably over the last two decades as new medications have been developed. When the number of drugs available for treating PAH was limited, it was common practice to start a patient on a single agent and then add additional therapy if the patient failed to improve. Rather than switching patients from one agent to another, physicians preferred to simply add a second class of drug that targeted a different biologic pathway and continue the original therapy that the patient was taking. This approach developed for several reasons. First, when a PAH patient does not improve on a given therapy, it is difficult to exclude the possibility that the treatment is still having some beneficial effect and that the patient’s disease will not worsen if the drug is removed. Second, the several months that most medications need to be effective would result in patients essentially starting over if they were switched from one drug to another. Finally, a variety of animal studies began to demonstrate the additive effects of some pulmonary vasodilator agents when used in combination to blunt the development of experimental pulmonary hypertension [25].
The evolution of clinical practice to use multiple drugs in combination for the treatment of PAH led to the organization of numerous clinical trials to examine the efficacy of combination therapy (Table 16.1). Unfortunately, few studies have examined the efficacy of combination therapy for PAH in a properly controlled randomized clinical trial. In order to evaluate the difference in efficacy between two drugs versus one drug, patients need to be randomized to one of the three arms: (1) drug A + placebo, (2) drug B + placebo, or (3) drug A + drug B (Fig. 16.2). Due to the large number of treatment-naïve patients needed for this approach, this type of study protocol has not been used until very recently. Instead, the vast majority of clinical trials evaluating combination therapy in PAH have used a protocol where patients who are clinically stable on drug A are randomized to drug A + drug B or drug A + placebo (Fig. 16.2). The limitation of this “add-on” approach is that the beneficial effect in the combination therapy group may result from drug B simply being superior to drug A rather than any additive or synergistic effects between the two drugs. In other words, the effect may have been the same if the patients were switched to drug B from drug A instead of adding drug B to drug A.
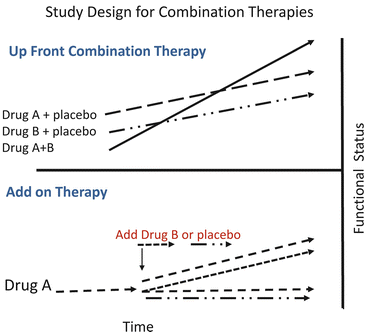
Table 16.1
Summary of major clinical trials of combination therapy
Study | Number enrolled | Study design | Treatment groups | Patient population | Primary outcomes | Results |
|---|---|---|---|---|---|---|
Phosphodiesterase inhibitors and prostacyclin analogues | ||||||
Ghofrani, H.A. et al. (2002) Ann Intern Med 136: 515–522. | 30 | Randomized, controlled open-label trial | 12.5 mg sildenafil, 50 mg sildenafil, 12.5 mg sildenafil + iloprost, 50 mg sildenafil + iloprost | Severe PAH or CTEPH | Maximum reduction of pulmonary vascular resistance (PVR), increase in cardiac index (CI) followed over 2 h | Iloprost + sildenafil more effective then sildenafil alone at reducing PVR and increasing CI |
Ghofrani, H.A. et al. (2003) J Am Coll Cardiol 42: 158–164. | 14 | Prospective non-randomized observational study | Patients who deteriorated on iloprost were given sildenafil + iloprost | Severe PPH, PAH | 6MWD over 12 months | Increase in 6MWD |
Simonneau, G. et al. (2008) Ann Intern Med 149: 521–530. | 267 | Double-blind placebo-controlled parallel group study | Patients being treated with epoprostenol randomized to sildenafil or placebo | Class I–IV PAH | 6MWD, mean PA pressure, cardiac output, time to clinical worsening over 16 weeks | Improvement in 6MWD, mean PA pressure, and cardiac output and time to clinical worsening |
Endothelin receptor antagonists and prostacyclin analogues | ||||||
Hoeper, M.M. et al. (2006) Eur Respir J 28: 691–694. | 40 | Multicenter randomized, open-label, controlled trial | Patients on bosentan randomized bosentan + inhaled iloprost or bosentan + placebo | Class III PPH | 6MWD over 12 weeks | No change in 6MWD |
Humbert, M. et al. (2004) Eur Respir J 24: 353–359. | 33 | Double-blind placebo-controlled prospective study | Patients starting epoprostenol randomized to bosentan or placebo in a 2:1 ratio | Class III or IV PAH | TPR, 6MWD, dyspnea score, NYHA functional class over 16 weeks | No change in TPR, dyspnea, 6MWD, NYHA class |
Mclaughlin, V. et al. (2006) Am J Respir Crit Care Med 174: 1257–1263. | 67 | Double-blind randomized multicenter trial | Patients stable on bosentan randomized to bosentan + iloprost or bosentan + placebo | Class III or IV PPH and PAH | 6MWD, change in NYHA class over 12 weeks | Increase in 6MWD and improvement in NYHA class |
Mclaughlin, V.V. et al. (2010) J Am Coll Cardiol 55: 1915–1922 | 235 | Randomized placebo-controlled multicenter study | Patients receiving a stable dose of sildenafil or bosentan randomized to treprostinil or placebo | Class III or IV PAH | 6MWD over 12 weeks | Significant improvement in 6MWD |
Seyfarth, H.J. et al. (2005) Chest 128: 709–713. | 16 | Prospective non-randomized open-label study | Patients on a background of beraprost or iloprost randomized to add bosentan or placebo | Class II–IV PAH or CTEPH | RV function index, 6MWD followed from 6 to 22 months | Improvement in RV function index and 6MWD |
Prostacyclins and endothelin receptor antagonists and/or phosphodiesterase-5 inhibitors | ||||||
Tapson, V.F. et al, (2012) Chest 142:1383–1390. | 350 | Double blind, placebo controlled | Patients on an ERA, PDE-5 inhibitor, or both, randomized to oral treprostinil initiated at an initial dose of 1 mg bid, or placebo | Severe PAH | Change in 6MWD from baseline to week 16 | No change in 6MWD |
Tapson, V.F. et al, (2013) Chest in press. | 310 | Double blind, placebo controlled | Patients on an ERA, PDE-5 inhibitor, or both, randomized to oral treprostinil initiated at an initial dose of 0.25 mg bid, or placebo | Severe PAH | Change in 6MWD from baseline to week 16 | No change in 6MWD |
Phosphodiesterase-5 inhibitors and endothelin receptor antagonists | ||||||
Hoeper, M.M. et al. (2004) Eur Respir J 24: 1007–1010. | 9 | Prospective non-randomized observational study | Patients on bosentan with clinical deterioration or no improvement were given sildenafil + bosentan | Class III or IV PAH | 6MWD over 12 weeks | Improvement in 6MWD and CPET |
Publication pending. | 500 | Randomized, double-blind, placebo-controlled trial | Treatment-naïve patients randomized to tadalafil 40 mg + placebo, ambrisentan 10 mg + placebo, or tadalafil 10 mg + ambrisentan 10 mg | Class III or IV | Time to clinical worsening | Time to clinical worsening was significantly longer in patients given tadalafil + ambrisentan compared to either drug alone |
Fig. 16.2
Proper testing of the efficacy of combination of two therapies requires three arms. One tests the efficacy of the first drug alone, the second arm tests the efficacy of the other agent alone, and the third arm tests the efficacy of the two drugs together (up-front combination therapy). In order to blind the patient and investigators a placebo for each drug is required. Studies that add either a second drug or placebo after patients have already been treated with another agent (add-on therapy) cannot determine if any improvement is due to the combination of the two medications or the second medication alone. See text for discussion
There is also a problem with selection bias in “add-on”-type trials. Patients may be more likely to be recruited into an “add-on” study if they had an unsatisfactory response to their initial therapy and this may increase the likelihood that they respond to the new drug. Such selection bias is shown in Fig. 16.3. If some PAH patients are more responsive to one class of PAH medication than the other drug classes, then monotherapy would select out those patients who do not respond to the initial medication. When a new medication is added to the failed background therapy, patients who improve may simply be responding to the new medication and not the combination of the original and newly added agent (Fig. 16.3a). If, however, the great majority of PAH patients have similar responses to all of the three major drug classes, then it is unlikely that the beneficial effect of adding a new medication to the old is due to the new medication alone. In this scenario, the beneficial treatment is likely to be due to the additive or synergistic effect of the two medications working together (Fig. 16.3b). Despite these limitations, a considerable body of data has been produced using add-on studies to examine the effect of combination therapies in PAH.
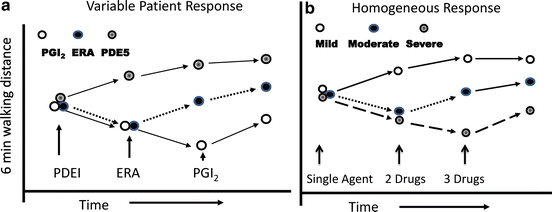
Fig. 16.3




Patient response to multiple therapies may be affected by how varied the underlying pathogenesis is in the treatment group. (a) If the study population consists of a mix of patients with different underlying mechanisms for their disease such that some are more likely to respond to one class of medication than the other, then clinical improvement observed after addition of a new therapy may be due to the new therapy alone. In this scenario, the benefit of multiple medications derives from increasing the odds that the patients will receive a medication that they are capable of responding to rather than any synergistic effects of the medicines used. (b) If all patients studied have the same pathogenic mechanism for their disease and are expected to respond similarly to each medication, then clinical improvement observed after addition of a new therapy to the old is more likely to be the result of the combined effect of the medications. In this example, patients with mild disease may only need one drug to improve while those with more severe disease require the additive or synergistic effects of multiple medications. PDEI phosphodiesterase inhibitor, ERA endothelin receptor antagonist, PGI 2 prostacyclin
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
