Fig. 6.1
Left panel: total occlusion of the left anterior descending (LAD) coronary artery which is completely reperfused by collateral vessels from the right coronary artery (RCA) in a patient with stable angina, normal LV function, no evidence of coronary-artery spasm and no other coronary stenoses. Right panel: during Holter ECG monitoring a large variability in ischemic threshold was observed, with transient myocardial ischemia (ST-segment depression) being often not observed at heart rate higher (bottom ECG strip) than that associated with ST-segment depression in other periods of the day (upper ECG strip).This large modulation of ischemic threshold during Holter ECG monitoring could result only from important vasomotor changes of the coronary microcirculation and/or collateral circulation. Adapted from Pupita et al. [8]
In patients with obstructive atherosclerosis of large epicardial arteries, the occurrence of myocardial ischemia during increased oxygen demand is usually attributed to the inadequate flow increase despite maximal vasodilatation of the coronary microvasculature [9]. According to this view, maximal coronary flow capacity and CFR have been proposed as accurate functional descriptors of stenosis severity to identify the need for revascularization [10]. Yet, the clinical correlation between stenosis severity and CFR is widely scattered [3, 4, 11–14]. These findings suggest that other factors might precipitate myocardial ischemia. For instance, the angiographic severity of coronary stenoses can increase during exercise or atrial pacing because of increased tone [15]. More importantly, in experimental studies, an increase in calculated total coronary resistance has been observed during tachycardia in regions supplied by severely stenotic coronary arteries [16]. Thus, it seems conceivable that in CAD, the response of the coronary microcirculation to increased oxygen demand might be more complex than a progressive arteriolar vasodilatation and that changes at the level of coronary stenosis may modulate flow response. To further clarify the possible role of the microcirculation, Sambuceti et al. assessed the resistance of both the stenotic arterial segment and the downstream microvasculature at rest and during pacing-induced ischemia in patients with stable angina. The authors found that tachycardia-induced ischemia was associated with an increase of the resistance of both the stenotic segment and the microvasculature. Interestingly, the infusion of adenosine during pacing reduced microvascular resistance (Fig. 6.2) [17]. This paradoxical behavior of vasomotor tone might reflect the intrinsic control mechanisms of the coronary circulation finalized to the maintenance of the driving pressure in a range of values high enough to perfuse vessels, but low enough to prevent capillary damage (see Chap. 1). Such a control could be as powerful as the metabolic control, although the two may go in opposite directions in this pathological condition. In this line, the response of coronary microcirculation to excessively low perfusion pressure could be a heterogeneous vasoconstriction, able to maintain pressure at the cost of excluding some vascular units. Although somewhat paradoxical and apparently not finalized to avoid or reduce ischemia, this hypothesis agrees with the evidence of both flow heterogeneity during hypoperfusion [18] and heterogeneous distribution of the metabolic fingerprints of ischemia in the subendocardial and subepicardial layers of the LV [19]. It is also worth mentioning that this complex microvascular response which is beneficial when microcirculation is intact, as it is the case in canine experimental models, might contribute to ischemia in atherosclerotic patients. Accordingly, in patients with severe epicardial coronary stenoses, adenosine administration during pacing resulted not only in reduction of microvascular resistance but also in reduction of myocardial ischemia, thus confirming that microvascular constriction contributes to negatively modulate the severity of ischemia [20].
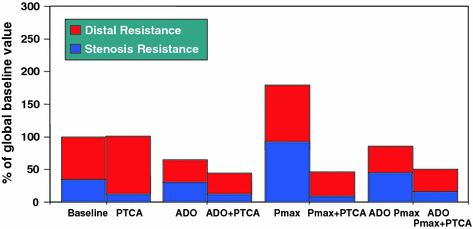
Fig. 6.2
Trend of stenosis, distal and global resistances expressed as percent of global baseline resistance. Stenosis resistance index was calculated as the ratio between mean trans-stenotic pressure gradient and blood flow index. Distal resistance index was calculated as the ratio between distal coronary pressure and flow index. Global resistance = distal + stenosis resistance. Intracoronary adenosine (ADO) markedly reduced distal resistance (p<0.05) and, to a lesser extent, stenosis resistance. During maximum atrial pacing (Pmax) an increase in both stenosis and distal resistance was observed (p<0.05 compared to baseline). Adenosine administered during atrial pacing markedly reduced distal and stenosis resistance compared to Pmax alone (p<0.05). After coronary angioplasty (PTCA), both stenosis and distal resistance remained low during Pmax. Adapted from Sambuceti et al. [17]
The “plaque-centric” hypothesis can also be called into question when the impact of therapeutic strategies based on the removal of coronary atherosclerotic obstructions is considered. Most reports agree that, on a background of medical therapy, revascularization improves symptoms, but in many patients angina recurs after 2–3 years and myocardial infarction and death are not prevented. In the COURAGE trial, which evaluated PCI on top of optimal medical therapy, more than 30 % of patients were still symptomatic with angina 1 year after the percutaneous procedure. Interestingly, at 5-year follow-up, the incidence of angina was not significantly different from that in patients who did not undergo a revascularization procedure (Fig. 6.3) [21]. Moreover, no significant between-group differences were noted for the composite end-point (death, myocardial infarction, and stroke), all-cause mortality, hospitalization for acute coronary syndrome, or myocardial infarction. Similarly, in the RITA-2 trial patients randomized to myocardial revascularization exhibited an initial greater improvement of symptoms and quality of life as compared to patients randomized to medical treatment. Yet, at 3-year follow-up, these differences between the two groups were not significant (Fig. 6.4) [22]. The pathophysiological relevance of obstructive lesions in the genesis of ischemia was further elucidated in the FAME study, where patients with multivessel coronary artery disease were randomly assigned to undergo PCI with implantation of DES guided by angiography alone or guided by FFR measurements in addition to angiography [23]. Patients assigned to FFR-guided PCI underwent stenting of indicated lesions only if the FFR was ≤0.80, whereas those assigned to angiography-guided PCI underwent stenting of all angiographically significant lesions. The 1-year event rate was significantly higher in the angiography-guided group than in the FFR-guided group (18.3 % vs. 13.2 %), in spite of a smaller number of stents implanted in the former. Importantly, 21 % of patients in the angiography-guided group and 19 % of patients in the FFR-guided group still complained of angina at 1-year follow-up, a figure similar to that observed in the COURAGE trial. The FAME 2 study randomized patients in whom at least one stenosis was functionally significant (FFR ≤0.80) to FFR-guided PCI plus optimal medical therapy or optimal medical therapy alone. Again the incidence of death and myocardial infarction was similar in the two groups. In contrast, the rate of urgent revascularization was higher in patients randomized to optimal medical treatment only and for this reason the study was closed prematurely. However, as FAME 2 was an open label study, a soft end-point like “urgent revascularization” is difficult to interpret [24].
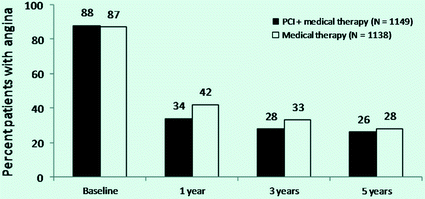
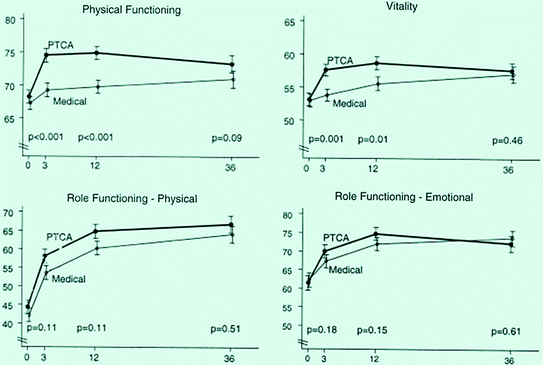
Fig. 6.3
Rate of angina after PCI in the COURAGE trial. Compared to baseline, angina rates decreased in both groups. Significant differences between treatment groups were noted at the 1-year and 3-year time points, but not at 5-year time. Adapted from Boden et al. [21]
Fig. 6.4
Comparisons of percutaneous transluminal coronary angioplasty (PTCA) and medical therapy in the RITA-2 trial: effects on different dimension of quality of life. The initial benefit observed with PTCA was not maintained at 3–6 year follow-up. Adapted from Pocock et al. [22]
In addition, in the STICH study, in patients with obstructive atherosclerosis and heart failure, no significant difference in all-cause mortality was observed between patients randomized to the medical therapy and those randomized to CABG [25]. Other large clinical trials have consistently shown a lack of mortality and symptomatic benefit in patients with stable angina and low to intermediate risk randomized to stent implantation in addition to optimal medical treatment or to optimal medical treatment alone [26] (Fig. 6.5).
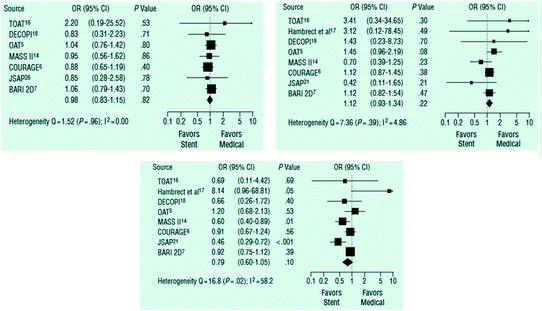
Fig. 6.5
Metanalysis of clinical trials that compared percutaneous coronary intervention with stent implantation with optimal medical therapy. Top left panel: death. Top right panel: non fatal AMI. Bottom graph: persistent angina. Adapted from Stergiopoulos et al. [26]
These disappointing results for symptom control in patients with stable angina after coronary revascularization have been attributed to a number of factors, including incomplete revascularization, in-stent restenosis, and patient-related factors. However, in a highly selected cohort of 220 patients in which all possible confounding factors had been excluded, one-third of patients were symptomatic with angina and presented with a positive exercise stress test 1 month after the index procedure [27].
These observations certainly do not deny the usefulness of revascularization in patients with ACS, when coronary revascularization is needed to prevent an impending catastrophe, but they do raise questions about whether revascularization should continue to be regarded as the ultimate treatment for obstructive atherosclerosis in stable patients. The unchanged prognosis and the high recurrence rate of angina clearly suggest that revascularization procedures remove the atherosclerotic obstructions but do not cure the underlying disease. Indeed, obstructive plaque is only one of the causes of stable angina, the other being represented by CMD.
As proof of concept, it is worth mentioning an elegant trial in which patients with stable angina and inducible myocardial ischemia were randomized to percutaneous coronary revascularization or a program of physical training. After 1-year follow-up, major cardiovascular event rate was lower and exercise capacity was better in the latter. This study highlights that physical training-related improvement of microvascular endothelial function and, perhaps, of collateral circulation function are more important than removal of epicardial coronary stenosis in improving symptoms and outcome in patients with stable angina [28].
Endothelial dysfunction has been shown to play an important role in the development of atherothrombosis through its regulation of vascular tone, platelet activity, leukocyte adhesion, and thrombosis [29]. Endothelial dysfunction is a predictor of cardiovascular events in patients with stable angina with or without obstructive atherosclerosis when assessed in the peripheral circulation, in large epicardial arteries and in coronary microcirculation [30, 31]. In keeping, coronary vasomotor response to acetylcholine has been shown to be an independent predictor of cardiovascular events in women with no, noncritical or critical epicardial stenosis, and suspected ischemia [32]. Accordingly, in a recent follow-up study of women with suspected myocardial ischemia, decreasing levels of CFR assessed by intracoronary adenosine (a nonendothelium dependent vasodilator stimulus) were significantly related to increasing risk of major adverse cardiovascular events (death or hospitalization for nonfatal AMI, congestive heart failure or stroke), with an adjusted hazards ratio of 1.14 per unit decrease in log-transformed CFR [33]. Inflammation is also a prominent part of the pathological process. Monocyte-derived macrophages and T-lymphocytes produce and secrete mediator molecules, such as cytokines, chemokines, growth-factors, proteolytic enzymes, and adhesion molecules, which activate endothelial cells, increase vasoreactivity, and cause smooth muscle cell proliferation. These events can enhance atherosclerotic plaque progression when occurring in large epicardial coronary arteries, while they cause dysfunction when occurring in coronary microcirculation by promoting both functional and structural alterations. In particular, plasma CRP, a prototypic marker of inflammation has been shown to be a risk factor for cardiovascular events in asymptomatic as well as in patients with known obstructive atherosclerosis [34, 35].
Women represent an interesting patient population in whom CMD is likely to play a key role in accounting for angina and myocardial ischemia in the presence of obstructive atherosclerosis [36]. In fact, while presenting with a lower coronary atherosclerotic burden, cardiovascular morbidity and mortality in women is similar to that in men [37]. The morbidity and mortality associated with the disease in this patient population have been a driving force in the exploration of other causes of myocardial ischemia including CMD. In line with these considerations, microvascular dysfunction in chronic angina can almost be considered as a “female gender disease.” According to experimental studies, sex plays a relevant role in a number of microvascular mechanisms which may affect microvascular function and disease. Sex-specific differences in microvascular blood flow and vasodilator capacity are observed very early in development.
In summary, a large body of evidence supports the notion that obstructive atherosclerosis is neither a sufficient nor a necessary cause for stable angina, but just one component of a complex pathophysiologic process. In patients with stable angina and obstructive atherosclerosis, the mechanisms responsible for angina are complex and related to both the presence of critical stenoses in large epicardial coronary arteries and the presence of microvascular dysfunction, which can also affect collateral circulation.
6.1.2 Clinical Implications
A microvascular origin of angina in patients with obstructive coronary atherosclerosis can be suspected in patients who have prolonged angina or angina poorly responsive to sublingual nitrates (clinical features frequently observed in patients with MVA). It can also be suspected in patients in whom angina is more severe than predicted by the severity of coronary stenoses. Finally, it may be suspected in patients in whom the angina threshold is remarkably variable, although this variability can also be accounted for by the presence of “dynamic” stenoses. In the individual patients, however, it is frequently impossible to establish the role played by CMD in causing angina. Therefore, myocardial revascularization should be considered in all patients at high risk (annual cardiovascular mortality rate >2 %) based on the results of stress testing and/or in patients with symptoms refractory to optimal anti-anginal medical treatment [38].
It is predictable, however, that up to 30 % of patients will have persistence of angina and/or evidence of stress-induced ischemia caused by CMD in spite of successful myocardial revascularization. Thus, when the goal of myocardial revascularization is symptom control rather than outcome improvement, it is always worth to test optimal anti-anginal treatment, including drugs targeting CMD, before proposing a procedure of myocardial revascularization.
The use of DES in PCIs has markedly increased during the past few years [39] mainly because they are associated with a significantly lower rate of restenosis compared to bare metal stents [40–42]. However, several studies have shown an increased risk of in-stent thrombosis associated with DES, which makes necessary the use of double anti-platelet drug therapy (aspirin and clopidogrel) for at least 6 months or more [43, 44]. Several factors have been associated with the enhanced prothrombotic effect of DES, including increased tissue factor expression [45] and impaired proliferation and migration of endothelial cells [46]. Accordingly, several studies have reported a significant coronary endothelial dysfunction associated with DES [47]. In particular, a recent study showed that endothelium-dependent vasomotion at adjacent stent segments was preserved after BMS implantation but not after DES implantation probably due to negative vascular effects of drug release [48].
It would be highly desirable to identify patients who have angina and inducible ischemia caused by CMD as opposed to that caused by restenosis. Exercise stress test has a primary role in the diagnosis and prognostic assessment of CAD [38]. However, the test performed after a PCI has been found of limited utility in identifying patients with coronary restenosis. In particular, previous studies showed that, after a percutaneous intervention, exercise-induced ST-segment depression at the ECG may persist, despite complete revascularization, in a proportion varying from 13 to 64 % [49–51].
The causes of ST-segment depression during exercise stress test in this setting, however, remain debatable. Although it is usually considered a false positive result, it might instead reflect myocardial ischemia caused by CMD [52, 53]. Recently, Milo et al. investigated patients with single-vessel LAD artery disease 24 h after successful stent implantation. All patients underwent exercise stress test and investigation of coronary microvascular dilatory function, assessed by measuring the changes in CBF velocity in the LAD coronary artery by TTDE in response to adenosine and to CPT. ST-segment depression during exercise stress test was found in about one-third of patients. No clinical or procedural significant differences were observed between patients with a positive and those with a negative result of exercise stress test. Notably, patients with a positive exercise stress test showed a lower CBF response to adenosine compared to those with negative exercise. At 3-month follow-up, 41 % of patients developed ST-segment depression during exercise stress test. Again, patients with positive exercise stress test showed a lower CBF response to adenosine compared to those with negative test. A low CBF response to adenosine (<2.0) was found in 82 % of patients with a positive test and in 18 % of those with a negative test. At 6-month follow-up, 45 % of patients developed ST-segment depression during exercise stress test. At this stage, a reduced CBF response to adenosine was present in all patients with a positive test whereas it could be detected in 6 % only of those with a negative test (Fig. 6.6). Of note, CBF response to CPT also was lower in patients with positive, compared to those with negative, exercise test up to 6-month follow-ip [54].
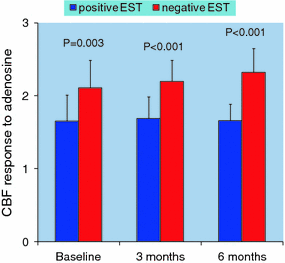
Fig. 6.6
Coronary blood flow (CBF) response to adenosine in patients with positive vs. negative exercise stress test (EST) 24 h (baseline) after a percutaneous coronary intervention and at 3-month and 6-month follow-up. Adapted from Milo et al. [54]
Finally, El Tamimi et al. [55] found that 50 % of patients treated by successful balloon angioplasty had a positive exercise stress test 1 week after the procedure; of note in all patients the test became negative after administration of sublingual nitrates. In the same patients intracoronary ergonovine failed to cause coronary spasm of epicardial arteries. They concluded that the cause of stress-induced ischemia was inappropriate vasoconstriction of coronary microcirculation.
The results of the above-mentioned studies do not allow us to discern whether CMD responsible for the positive response to exercise stress testing early after successful angioplasty was caused or just unmasked by the procedure. Furthermore, the results of these studies clarify that a positive result of an exercise stress test can be caused by either restenosis or by CMD and that no distinctive feature can separate between these two possibilities, although a positive result early after stent implantation is likely to be caused by CMD, while a negative test which becomes positive after months is more likely to reflect restenosis, although progressive microvascular dysfunction cannot be excluded. Imaging techniques as well appear inadequate to discriminate between CMD and restenosis. In one study in patients with stable angina and one-vessel disease who had undergone successful stent implantation, early adenosine thallium-201 scintigraphy showed transient perfusion normalities in the region supplied by the treated coronary artery branch in about half of patients [56].
A more direct noninvasive assessment of coronary anatomy would be desirable in this setting, which might be achieved by multislice computed tomography coronary angiography. Yet, this technique still has important limitations in the assessment of in-stent restensosis; furthermore, it is associated with a significant radiation exposure and does not allow assessment of functional coronary abnormalities.
Thus, with the exception of invasive coronary angiography, all noninvasive techniques are unable to reliably discriminate between angina and myocardial ischemia caused by CMD vs restenosis.
Taken together, a large body of evidence gathered in the past 10–15 years indicates that among patients with stable angina both symptoms and myocardial ischemia are caused by both epicardial stenoses and CMD: their relative clinical relevance probably varies in different patients and varies over time in the same patient. In patients in whom myocardial revascularization is not dictated by a poor outcome related to a large area of myocardium at risk of ischemia, but rather by the need to improve invalidating anginal symptoms, the main goal is optimal medical anti-anginal treatment based on drugs which reduce myocardial oxygen demand and improve myocardial perfusion operating at the site of both large epicardial vessels and coronary microcirculation. Lack of appreciation that symptoms can originate from CMD can expose to unnecessary revascularization procedures. This is particularly important in patients who have persistence of symptoms and/or of evidence of inducible ischemia after successful myocardial revascularization.
Because of the lack of information on how to identify the contribution of epicardial stenoses and of CMD to anginal symptoms in the single patient, in patients with stable angina who do not have a large area at risk of myocardial ischemia, the main goal of treatment should be global risk factor control and anti-anginal treatment targeting both large epicardial coronary arteries and coronary microcirculation.
6.2 Acute Coronary Syndromes
6.2.1 Pathophysiology of CMD
The pathophysiology of CMD in ACS has not fully been elucidated. In the vast majority of patients, ischemic attacks are associated with reduction in CBF and occlusion of a major coronary branch attributed to thrombosis.
In contrast, coronary spasm due to smooth muscle hyperreactivity is the predominant cause of myocardial infarction in patients with a history of vasospastic angina, although this event is less common. However, coronary vasoconstriction and thrombosis are deeply interrelated [57–59].
On the one hand, occlusive coronary spasm and distal blood stagnation are known to cause a transient several-fold increase of fibrinopeptide A in systemic blood [60, 61]. On the other hand, serotonin, a substance released by activated platelets, is known to produce occlusive spasm in patients with variant angina and myocardial ischemia caused by distal vessel constriction in patients with chronic stable angina [62]. This vicious cycle, at least partially, mediated by serotonin, thromboxane A2, and thrombin, may have an important role in the setting of ACS where unstable coronary plaques, frequently showing a preserved smooth muscle, are in contact with activated platelets [63]. Furthermore, several findings sustain the possibility of smooth muscle hyperreactivity in unstable angina. Indeed, ergonovine injection causes occlusive spasm much more often in unstable than in stable angina (38 % vs. 4 %), a difference similar to that observed in patients with recent infarction compared with patients with old infarction (20 % vs. 6 %) [59]. It is also known that unstable plaques are more reactive to the stimuli of exercise and CPT than stable plaques [64]. Furthermore, Zeiher et al. [65] have demonstrated greater endothelin-1 immunoreactivity in unstable coronary plaques compared with that found in stable plaques obtained by directional atherectomy. This observation may provide a clue to the mechanisms of segmental coronary hyperreactivity frequently observed in patients with unstable angina [64, 65]. Indeed, endothelin-1 is not only a potent vasoconstrictor itself, but it also potentiates the effects of other vasoconstrictor stimuli such as catecholamines, serotonin, and angiotensin II [66].
A growing body of data indicates that plaque inflammation may play a key role in the pathogenesis of ACSs, as the cytokines secreted by activated inflammatory cells have the potential to activate the endothelium, transforming its antiadhesive and anticoagulant properties into adhesive and procoagulant properties. Furthermore, they may reduce matrix synthesis and increase its degradation, thus favoring plaque rupture. Finally, they may increase endothelin-1 synthesis in endothelial cells and macrophages and endothelin-1, in turn, can enhance smooth muscle reactivity to other constrictors. Hence, plaque inflammation may account for the multiple mechanisms so far proposed as being directly responsible for ACSs.
Thus, over the last two decades, the working hypothesis has been that ACSs are primarily an epicardial or a large vessel event as a result of plaque rupture or erosion [67]. At the time of infarction, the angiographic display of an occluded epicardial artery with intra-luminal thrombus and slow or even absent coronary flow that persists after recanalization led to the widely accepted opinion that the coronary microcirculation is an innocent bystander in the initial phase of the acute coronary event [68].
According to these classical concepts, a sudden focal increases in epicardial coronary artery resistance should be associated with compensatory vasodilatation at the level of microcirculation [69]. However, experimental studies have shown that this is not the case during prolonged severe ischemia [70, 16]. Moreover, clinical studies suggest the occurrence of a microvascular vasoconstriction during transient myocardial ischemia in patients with stable angina [20, 71] and Wilson et al. observed intense symptomatic microvascular constriction after angioplasty of acute thrombotic coronary lesions [72]. Accordingly, elegant studies documented that substances released during platelet aggregation can constrict coronary microvasculature, particularly in presence of atherosclerotic endothelial dysfunction [73]. These observations emphasize the need for a better understanding of the role of microcirculatory vasomotor tone during ACS. To this purpose, simultaneous evaluation of blood flow changes together with proximal and distal coronary pressures are required. This information can be obtained in humans by measuring CBF velocity using an intracoronary Doppler wire and a pressure wire with the positioned distal to the culprit stenosis [74]. Using this approach, Marzilli et al. found that, in these patients, transient myocardial ischemia was associated with paradoxical vasoconstriction of both stenotic arterial segment and downstream microcirculation [75] (Fig. 6.7). These findings do not agree with the traditional view of a maximal compensatory vasodilatation in ischemic myocardium. By contrast, they strongly support the hypothesis that spontaneous ischemia can be associated with microvascular constriction and suggest that abnormalities in distal coronary vasomotion can contribute to the precipitation and maintenance of ischemia in unstable angina. Along this line, intermittent ischemia might reflect mural thrombosis and release of substances able to constrict coronary microvessels [76, 77]. Alternatively, vasoactive signals not linked to coronary thrombosis might be responsible for coronary vasoconstriction. Actually, endothelial dysfunction impairs microvascular adaptation to ischemia [78], and a constrictor response to reduced intraluminal pressure has been described in isolated microvessels [79]. Similarly, ischemia mediated by stimulation of postsynaptic alpha-2-receptors has been documented in anesthetized dogs [80]. Potent coronary vasoconstrictors such as neuropeptide-Y [81] and endothelin-1 [82] are more effective on coronary microcirculation than on large epicardial arteries arteries.
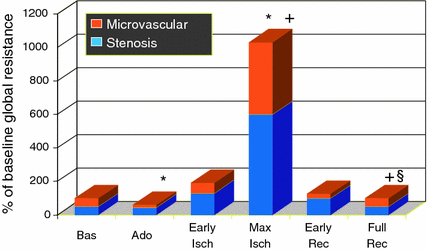
Fig. 6.7
Trends of total coronary resistance and the contribution of stenosis (blue columns) and microvascular resistance (orange columns). Coronary resistance was not calculated during balloon coronary occlusion. During ischemia, both coronary stenosis and distal microcirculation showed a significant increase in resistance to flow (*p < 0.05 vs. baseline, +p < 0.05 vs. adenosine; § 5 p < 0.05 vs. max ischemia). Adapted from Marzilli et al. [75]
Identifying the mechanisms of the altered control of vasomotor tone, during ischemia, might allow a more accurate identification of patients who will develop an acute coronary disease and the development of more specific forms of anti-ischemic therapy. At present, the most used vasoactive drugs are most effective on large epicardial arteries; in fact, direct arteriolar vasodilators may precipitate ischemia by coronary steal. The identification of the mechanisms underlying this paradoxical microvascular constriction and thus of specific substances able to prevent it, might provide a more efficient pharmacologic strategy especially for those patients who cannot benefit from coronary revascularization.
Interestingly, Marzilli et al. found that abciximab, a GP IIb/IIIa antagonist, immediately improved MBF in patients with ACSs, mostly because of the beneficial effect on coronary microcirculation (Fig. 6.8) [83]. In agreement with other published reports, these data suggest that GP IIb/IIIa antagonism might prevent MVO, thus increasing the amount of myocardial mass reached by perfusion. These observations offer an alternative explanation for the clinical benefits of anti-platelet agents in patients with ACS.
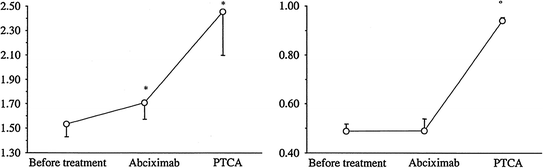
Fig. 6.8
The coronary flow reserve (CFR) (left panel) and fractional flow reserve (FFR) (right panel) responses to abciximab and percutaneous transluminal coronary angioplasty (PTCA). Abciximab improved CFR but not FFR, indicating a greater effect of the drug on coronary microvascular function than on stenosis severity. Data are shown as average values SEM. *p 0.05 vs. baseline. °p 0.01 vs. baseline and abciximab. Adapted from Marzilli et al. [83]
Taken together, these data suggest that, besides changes in plaque structure and morphology, microvascular phenomena might contribute to precipitating ischemia in unstable patients. Although the mechanism(s) of the paradoxical constriction of the coronary microcirculation during ischemia are largely unknown, some reports suggest that platelets might interfere with microvascular regulation of CBF. Platelets may be activated by exposure to thrombus, injured endothelium, and collagen while crossing the stenotic segment [84]. Activated platelets can affect microvascular resistance by microembolization and/or release of constrictive, pro-adhesive, and pro-inflammatory factors. Along this line, ACSs are often associated with evidence of microvascular obstruction [85]. Although this phenomenon is generally attributed to microembolization from the epicardial thrombus, it has been documented that interaction with adhesion molecules prevents MVO in experimental models of ischemia without coronary thrombosis [86].
Thus, at the time of an acute coronary event: (1) CMD, characterized by paradoxical vasoconstriction, seems to play an important modulatory role; (2) anti-platelet drugs known to improve clinical outcome appear to improve CMD.
6.2.2 Relations Between CMD and Plaques Instability
The temporal association between events occurring in large epicardial vessels (plaque erosion or fissure associated with thrombus formation) and in the microcirculation (paradoxical vasoconstriction) does not allow to establish what is the causal relation between these two events. It is widely believed that epicardial events precede and cause microvascular events. Yet this assumption is based on studies that were not designed to answer this question and is challenged by several clinical observations. Indeed, it has to be taken into consideration that plaque ruptures can remain clinically silent. Although the exact prevalence is unknown, autopsy studies do outline that healed ruptures are not infrequently encountered upon histopathological review of coronary arteries of sudden cardiac death patients [87, 88]. In fact, in one intravascular ultrasound-based study, almost one quarter of all ruptured plaques was encountered in patients with stable angina or no symptoms at all [89] and similar findings have recently confirmed using optical coherence tomography [90]. Moreover, by serial coronary angiography, one study suggested a delay of at least 3 days from plaque rupture and thrombus formation to the classical clinical presentation of acute myocardial infarction [91]. Furthermore, patients with similar coronary “patho-anatomy” may present with unstable angina rather than acute myocardial infarction or even sudden cardiac death. Hence, there is a fairly broad spectrum in the clinical presentation of an epicardial event, which has remained largely unexplained. While systemic factors such as thrombotic-fibrinolytic balance and local factors such as collateral blood flow have to be taken into consideration, one may also want to question the coronary microcirculation as a potential modulating factor.
Thus, one may postulate the presence not only of the vulnerable plaque but also of the vulnerable coronary microcirculation. A more extreme view is that, at least in a proportion of patients, a primary CMD might play a causal role in determining thrombus formation in epicardial coronary arteries. Several lines of evidence support this working hypothesis.
For instance, the fact that CBF is reduced by 50 % in the nonculprit coronary arteries in patients with acute myocardial infarction both before and after a PCI points to global rather than regional myocardial microcirculatory impairment [92].
Moreover, in patients without obstructive CAD and evidence of coronary epicardial and microvascular endothelial dysfunction during invasive cardiac evaluation, future cardiovascular events, including ACS, occurred more frequently in those with a reduction of CBF in response to vasodilator stimuli, indicating CMD [93]. Thus, Britten et al. who followed patients with angiographically normal or minimally diseased coronary arteries over an average of 6.5 years, noted a more than threefold higher cardiovascular event rate in patients in the lowest compared with the highest tertile of CFR with about one-third of all events related to ACSs [94]. Marks et al. followed patients with chest pain and normal coronary angiograms over a mean period of 8.5 years and noted a nearly threefold higher mortality for those patients with an abnormal CFR [95]. Hence, the presence of CMD is a significant predictor of clinical outcome, including future acute coronary events, even in the absence of hemodynamically significant epicardial disease [96]. These findings have recently been confirmed in a large cohort study of 11,223 Danish patients referred for coronary angiography because of stable angina pectoris and 5705 asymptomatic patients found to have angiographically normal coronary arteries Main outcome measures were major adverse cardiovascular events, defined as cardiovascular death, myocardial infarction, stroke or heart failure, and all-cause mortality. Significantly, more symptomatic women (65 %) than men (32 %) had non obstructive CAD. Notably, the multivariable adjusted risk of major cardiovascular events was 52 % higher for symptomatic patients with angiographically normal coronary arteries and 85 % higher for patients with diffuse non obstructive CAD compared with the reference population. In particular, the risk of all-cause mortality was 29 and 52 % higher, respectively [97]. Similar findings were observed in another study which compared clinical outcome of 540 women with non obstructive CAD enrolled in the WISE study (symptomatic women referred for clinically indicated coronary angiography because of angina or suspected ischemia) with those of 1,000 age and race-matched women enrolled in the WTH Project (asymptomatic, community-based women with no history of heart disease). Five-year annualised event rates for cardiovascular events were 16.0 % in WISE women with nonobstructive coronary disease, 7.9 % in WISE women with normal coronary arteries, and 2.4 % in asymptomatic WTH women after adjusting for baseline risk factors [98].
PCI can be considered as a iatrogenic form of plaque rupture and allows assessment of the significance of the myocardial microcirculation in a more defined setting. In this regard, it is important to notice that clinical studies of PCI pointed out that patients with pre-procedural CFR impairment were more likely to have post-procedural CFR impairment and procedure-related myocardial injury as well as worse long-term outcome [99, 100]. These data suggest that pre-existing impairment of coronary microcirculation yields greater vulnerability to myocardial injury and thereby imply a primary role of CMD, at least in PCI-related myocardial injury.
Inflammation may be a common link between macrovascular and microvascular disease. Indeed, Neri Serneri et al. demonstrated an acute inflammatory process involving coronary microvessels but not cardiomyocytes in unstable angina patients [101]. Accordingly, in patients with normal coronary angiograms, a significant inverse correlation was noted between CRP serum concentrations and MBF responses to cold pressor test measured by PET [102]. Based on these latter findings, one could plausibly speculate that the inflammatory mechanisms linked to plaque rupture also cause CMD.
Further support to this argument may be derived from previous retrospective and prospective randomized trials, which demonstrated a reduction in the incidence and extent of myocardial injury with PCI by pre-treatment with statins, known to improve microvascular function [103–105]. Even more, a recently published study highlighted a 74 % reduction in the incidence of MVO with primary PCI among patients who were taking statins before admission for anterior acute myocardial infarction, suggesting that chronic statin treatment could preserve the microvascular integrity [106] (see Chaps. 10 and 11).
Taken together, there is a growing body of multilayered evidence to suggest that the integrity of the coronary microcirculation plays an integral role in the evolution of ACS. In line with the concept of the primary significance of the myocardial microcirculation, pre-existing transient or permanent microcirculation dysfunction may contribute to the development and prognosis of ACS via reduction of CBF, leading to an alteration of shear stress and thereby aggravation of endothelial dysfunction in epicardial level as well as aggravation of thrombus formation in an extension of ‘Folts’ coronary thrombosis model [107]. Coronary thrombus formation as a primary or secondary event in acute myocardial infarction has been debated quite extensively in the past, and some authors would argue that reduction of flow is the primary factor and intracoronary thrombus develops after the onset of myocardial ischemia (Fig. 6.9) [108].
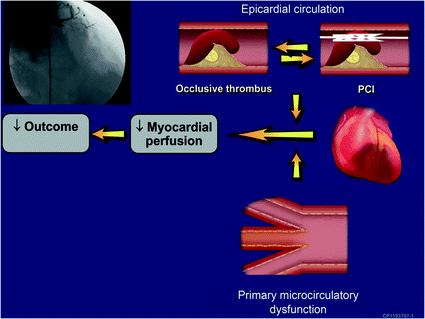
Fig. 6.9
A nontraditional concept of the mechanisms of ACS centers on the vulnerable patient, taking into account primary dysfunction of the microcirculation of a vulnerable myocardium in addition to the vulnerable plaque, which contributes to the clinical presentation and outcome. Adapted from Lerman et al. [108]
A limitation of the working hypothesis that CMD may play a primary role in the pathogensis of ACS relates to the question how microvascular function and basal CBF could deteriorate so acutely and so extensively to contribute to acute coronary thrombosis, especially because the majority of patients with acute myocardial infarction apparently do not display CMD at the time of assessment. Yet, acute deterioration in microvascular function and myocardial perfusion has been demonstrated in diabetic patients postprandially [109]. A dysfunctional endothelium is also increasingly sensitive to catecholamines, which may explain the sudden increase in cardiac events with stressful life events [110]. Accordingly, in healthy individuals, endothelium-dependent vasodilatation by forearm venous occlusion plethymography is lower in the evening than in the morning hours and thought to counteract potentially harmful diurnal patterns of sympathetic tone and coagulability [111]. However, in patients with angiographically proven obstructive CAD, this diurnal variation in endothelium-dependent vasodilatation was noted to be absent, conferring a potentially greater risk [111]. Finally, an excellent example of sudden intense microvascular constriction able to cause severe myocardial dysfunction in the absence of obstructive coronary atherosclerosis is represented by the takotsubo syndrome which is discussed elsewhere (see Chap. 4).
Although the hypothesis of a primary role of CMD is fascinating, a major limitation to argue for or against this notion is the lack of tracing data on microvascular function and CBF just prior to the acute coronary event. Thus, future studies should take these possibilities into account in the design of a novel clinical approach for the treatment of ACS. If indeed microcirculatory dysfunction is ever demonstrated to be one of the major contributors to the evolution and not just the consequence of an acute coronary event, this could substantially alter future research directions and approaches to therapy. The paradigm that an ACS may be initiated at the level of coronary microcirculation and results in thrombus formation at a site of a nonocclusive epicardial artery may not, however, change clinical practice, which is to restore CBF and myocardial perfusion. Nonetheless, it could exert an influence upon the selection of adjuvant therapy prior and/or following the revascularization procedure or even a novel design for DES.
6.3 Coronary Microvascular Obstruction
6.3.1 Definition and Clinical Relevance
In the setting of STEMI, a specific pathophsysiological condition is represented by CMD, which occurs after recanalization of the infarct-related artery. Prompt referral for mechanical reperfusion by urgent primary PCI represents the pivotal step in the current management of STEMI [112]. Yet, in a sizable proportion of patients primary PCI achieves epicardial coronary artery recanalization, but not myocardial reperfusion, a condition known as “no-reflow” and currently more commonly defined MVO [113].
Although the existence of MVO was initially debated, a large amount of experimental and clinical data has nowadays convincingly shown that it occurs after reperfusion with a variable prevalence, ranging from 5 % to 50 %, according to the methods used for the diagnosis and the population under study [114].
In 1993, at the climax of the thrombolytic era, Lincoff and Topol wrote a provocative editorial wondering whether reperfusion was just an illusion [115]. At that time, they estimated that only “25 % or less” of patients treated by thrombolysis had an optimal reperfusion, defined as a rapid, complete, and sustained coronary recanalization with adequate myocardial tissue perfusion. What is this figure after 15 years at the climax of primary PCI era? As shown in Fig. 6.10, a reasonable estimate of the proportion of patients who get optimal myocardial reperfusion, among those without cardiogenic shock undergoing primary PCI, is about 35 % [116].
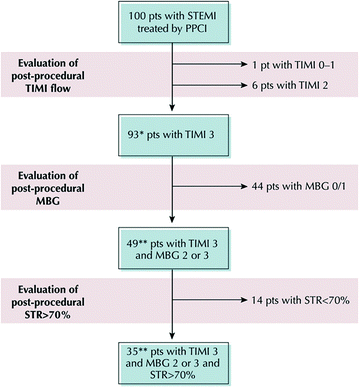
Fig. 6.10
Prevalence of myocardial MVO. Estimate of the number of patients (pts) receiving optimal reperfusion according to Thrombolysis In Myocardial Infarction (TIMI) flow grade, myocardial blush grade (MBG), and ST-segment resolution (STR) of 100 patients without cardiogenic shock treated by primary percutaneous coronary intervention (PPCI). *Estimation derived from 20 randomized trials comparing standard percutaneous coronary intervention with thrombectomy or distal protection. **Estimation derived from core laboratory analysis of the CADILLAC trial. Adapted from Niccoli et al. [116]
A series of consistent data have clearly shown that MVO has a strong negative impact on outcome, negating the potential benefit of primary PCI [117–122]. Indeed, patients with MVO exhibit a higher prevalence of: (1) early post-infarction complications (arrhythmias, pericardial effusion, cardiac tamponade, early congestive heart failure); (2) left adverse ventricular remodeling; (3) late re-hospitalizations for heart failure; and (4) mortality (Fig. 6.11).
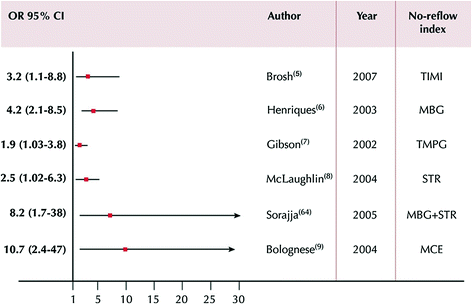
Fig. 6.11
Prognostic value of MVO according to angiographic, electrocardiographic, and echocontrastographic indexes. The end point was cardiac death or total mortality. Data are presented as odds ratio (OR) and 95 % confidence interval. Adapted from Niccoli et al. [116]
Therefore, detection, prevention, and treatment of MVO are likely to have an important impact on the outcome of primary PCI.
6.3.2 Time-Course and Pathogenetic Components
Kloner et al. described MVO for the first time in a canine model, demonstrating that it occurs after prolonged (90 min) coronary occlusion followed by reperfusion [123]. The consequences of coronary ligation of a non atherosclerotic coronary artery, however, cannot be directly extrapolated to the human situation where myocardial infarction is caused by occlusive coronary thrombosis superimposed to an atherosclerotic unstable plaque [124].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


