Fig. 5.1
Typical asymmetric LVH, which dramatically involves the interventricular septum, in a patients with HCM deceased due to sudden death. Adapted from Cecchi et al. [15]
Many patients with HCM remain asymptomatic or develop mild symptoms during life, and mortality rate is about 1 % per year, which is not significantly different from that expected in the general adult population. However, HCM can result in major cardiac events, including sudden death and heart failure.
Sudden death is a particularly dramatic event in HCM, as it usually hits young patients. HCM is indeed a major cause of sudden death occurring in athletes during sports’ activities. The mechanism of sudden death is ventricular tachycardia/fibrillation, and ICD implantation significantly improves survival [6].
Symptoms of heart failure occur in 10–20 % of cases and are more frequent in patients with LV outflow gradient >30 mmHg, significant diastolic dysfunction or development of atrial fibrillation, which also increases the risk of thromboembolic events. Evolution toward a phenotype similar to that of dilated cardiomyopathy occurs in about 5 % of patients and is only predicted by a family history of this complication.
Since its original description, myocardial ischemia has been a recognized, but underappreciated aspect of the pathophysiology of HCM. Symptoms and signs of myocardial ischemia are indeed often found in these patients, despite the usual presence of angiographically normal coronary arteries. Perfusion defects on myocardial thallium-201 single photon emission computed tomography and lactate production in the coronary sinus during pacing stress were indeed convincingly shown in HCM patients many years ago [7].
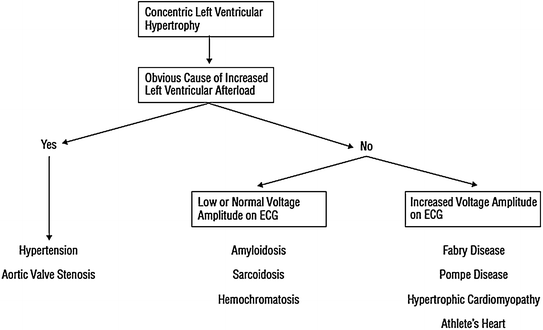
Moreover, in the past 20 years a number of studies [8–11] have demonstrated that CFR is severely blunted in these patients due to CMD (Fig. 5.2). Notably, CFR impairment is not confined to the hypertrophied interventricular septum, but it concerns also the less hypertrophied LV free wall, in line with the evidence of widespread remodeling of intramural arterioles at autopsy [12, 13].
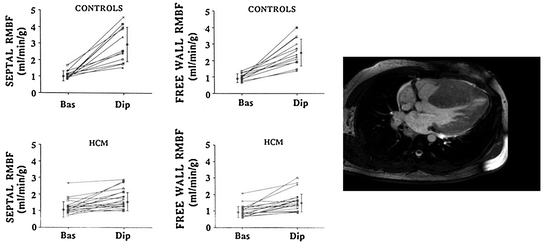
Fig. 5.2
Evidence for a significant reduction of hyperemic MBF (dipyridamole, Dip) in all myocardial regions (septum and LV free wall) in patients with HCM compared to normal controls. hRMBF=regional MBF Adapted from Camici et al. [12]
Notwithstanding the large evidence of data supporting an important pathophysiological role for CMD, the assessment of myocardial ischemia is currently not part of routine clinical diagnostic or management strategies in these patients, which represents a limitation that can significantly impact our capability to achieve a comprehensive evaluation of individual patients with HCM [14]. The severity of CMD in affected patients has indeed been found to be an independent predictor of long-term clinical deterioration and death from cardiovascular causes (Fig. 5.3) [15, 16].
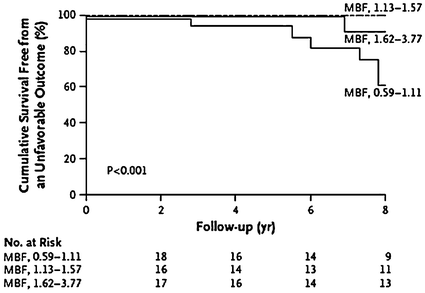
Fig. 5.3
Survival curves of patients with HCM according to hyperemic MBF. Prognosis was significantly worse in those with reduced hyperemic MBF indicating CMD. Adapted from Cecchi et al. [15]
Repeated episodes of focal myocardial ischemia, caused by CMD, might result in a progressive loss of cardiomyocytes and fibrotic replacement of the myocardium [17], which can significantly contribute to some of the severe complications of HCM, including ventricular arrhythmias and sudden death, as well as progressive LV remodeling and systolic dysfunction [18–21], eventually resulting in overt LV dysfunction and failure (Fig. 5.4) [22].
Fig. 5.4
Schematic representation of the relation between CMD and development of myocardial fibrosis, with evolution toward impaired LV function and arrhythmias, in patients with HCM. Adapted from Camici et al. [24]
More recently, several studies using CMR have led to an enhanced understanding of the role that myocardial ischemia and the resulting myocardial fibrosis may play on clinical outcome in patients with HCM. These studies have indeed demonstrated that late gadolinium enhancement, a reliable marker for myocardial fibrosis, at CMR has an outstanding prognostic value in predicting adverse cardiovascular events in patients with HCM. This has also been confirmed by a recent meta-analysis in over one thousand HCM patients, which has shown that late gadolinium enhancement significantly correlated, at an average follow-up of more than 3 years, with the development of heart failure and with all-cause and cardiac mortality, with a trend toward significance also for prediction of sudden death [21, 23–26] (Fig. 5.5).
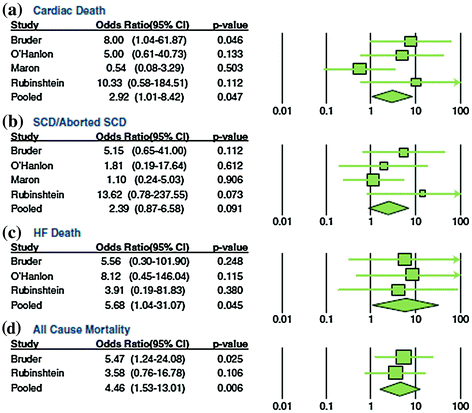
Fig. 5.5
Meta-analysis forrest plots and pooled odds ratios for clinical endpoints in HCM studies assessing the prognostic role of late gadolinium enhancement at CMR. The latter significantly predicted cardiac death, heart failure (HF) death, and all-cause mortality. Furthermore, a trend toward significance was observed for sudden cardiac death (SCD). Adapted from Green et al. [23]
The presence of sparse areas of fibrosis in the myocardium may indeed favor electrical instability and induction of life-threatening ventricular arrhythmias (see Chap. 4). Based on these studies, extensive late gadolinium enhancement at CMR is now considered among the features that indicate increased risk of sudden death in HCM, together with the classical predictors of familial history of sudden death, unexplained syncope, LV wall thickness >30 mm, recurrent episodes of non sustained ventricular tachycardia during ambulatory ECG monitoring and hypotensive response or attenuated increase in blood pressure during exercise [27].
It is worth noting that recent data indicate that [28] patients with evidence of sarcomere myofilament mutations are characterized by more severe CMD and an increased prevalence of myocardial fibrosis, compared with genotype-negative individuals, suggesting a direct link between sarcomere gene mutations and adverse remodeling of the microcirculation, accounting for the increased long-term prevalence of LV dysfunction and heart failure, and possibly arrhythmic events, in genotype positive patients.
5.2 Dilated Cardiomyopathy
Idiopathic dilated cardiomyopathy is a cardiac muscle disease characterized by reduced contractile systolic function with dilatation of the LV or, in some cases, of both ventricles [29]. Dilated cardiomyopathy is a major cause of heart failure, presenting an incidence of about 7 per 100,000 individuals [30]. Despite progress in medical therapy, prognosis remains unfavorable; mortality is indeed 20 % at 5 years [31], with worsening heart failure and ventricular tachyarrhythmias being the most frequent causes of death.
The specific causes of dilated cardiomyopathy remain largely unknown, and include genetic, viral, and autoimmune mechanisms [32]. In particular, an immunological reaction to an initial viral myocarditis, independently of virus persistence in cardiomyocytes, is believed a major cause of the disease, although the hypothesis remains to be proven [33].
The presence of CMD in a significant proportion of patients with dilated cardiomyopathy is suggested by the frequent occurrence of angina, which can sometimes precede the manifestation of symptoms and/or signs of heart failure, and is also supported by the detection of myocardial perfusion defects and/or regional LV wall motion abnormalities during stress tests, despite the evidence of normal coronary arteries at angiography [34].
A severely reduced CFR in patients with dilated cardiomyopathy has been demonstrated in several studies [35–39], and other studies have also shown the presence of impaired endothelium-dependent coronary microvascular dilatation [40].
Interestingly, CMD and MBF abnormalities can be diffuse or mainly localized in some heart regions, which, at PET studies, can also present increased anaerobic glucose metabolism [41, 42] (Fig. 5.6), similarly to the flow-metabolism “mismatch”, shown in myocardial “hibernation” of CAD patients, which represents the adaptation of the myocardial tissue to chronic or repetitive ischemia [43]. The question remains open, however, whether CMD is part of the disease or arises late as a consequence of heart failure. In fact, several mechanisms can be involved in determining CMD in dilated cardiomyopathy, including alterations of the coronary microcirculation secondary to significant eccentric LVH and/or dilatation, extravascular compression due to increased LV filling pressure, and MBF downregulation, due to reduced energetic demand [35]. Furthermore, despite the evidence of functional coronary microvascular abnormalities, no structural abnormalities of arteriolar vessels have usually been found in these patients, although recent data suggest possible changes in arterioles below 50 μm in diameter.
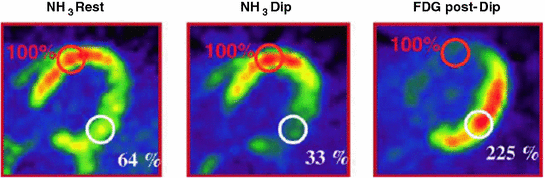
Fig. 5.6
PET scans of MBF and metabolism in a patient with early dilated cardiomyopathy. At baseline (NH3 rest) there is evidence of reduced tracer uptake (64 % of maximum) in the posterolateral wall of the LV; this MBF defect becomes more severe (33 % of maximum) during pharmacologic stress with dipyridamole (NH3 Dip). The 18F-fluorodeoxyglucose (FDG) scan shows increased glucose utilization uptake in the hypoperfused region. In the absence of obstructive CAD, this flow/metabolism mismatch suggests myocardial ischemia related to CMD. Adapted from Neglia et al. [41]
Yet, several findings suggest that small coronary arteries may present functional alterations with a potential causal role in determining the cardiomyopathy, at least in a subset of patients. A reduced CFR can indeed be detected in up to 82 % of patients with early dilated cardiomyopathy (i.e., without any sign of heart failure) [36]. Furthermore, no correlation has been found between impaired peripheral microvascular endothelial function and endothelial CMD, suggesting that the latter can be largely independent of the hemodynamic effects of heart failure [44]; finally, the impairment of MBF in these patients was found to be independent of the extension of myocardial fibrosis, as assessed in explanted hearts of patients with dilated cardiomyopathy [45].
Regardless of whether CMD is a pathogenetic component of the disease or is just a consequence of the abnormal cardiac or systemic effects of the disease, its occurrence can significantly contribute to the evolution of the disease by inducing foci of myocardial ischemia. Accordingly, repeated myocardial ischemia due to CMD might result in areas of necrosis and myocardial fibrosis, which might increase the risk of both progressive myocardial dysfunction and heart failure, but also of electrical instability and sudden death, as discussed above for patients with HCM.
As in the latter patients, indeed, also in those with dilated cardiomyopathy the degree of CMD has been shown to be an independent predictor of cardiac events and is associated with an increased relative risk of death and further progression of heart failure (Fig. 5.7) [46].
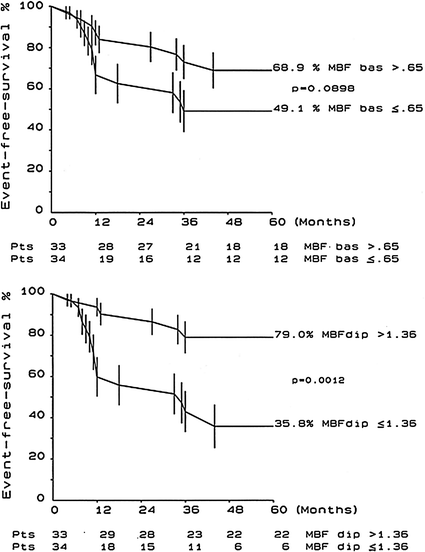
Fig. 5.7
Kaplan-Meier event-free survival plots at 5 years are shown for patients with dilated cardiomyopathy subdivided into groups according to the median value of resting MBF (MBF bas) (top panel) or dipyridamole MBF (MBF dip) (bottom panel). Adapted from Neglia et al. [46]
A number of clinical parameters are used for risk stratification of patients with dilated cardiomyopathy, including history of syncope, detection of ventricular tachycardia on ambulatory ECG monitoring, induction of ventricular tachyarrhythmias by programmed ventricular stimulation, QRS duration, and several other ECG derived indices, including heart rate variability, baroreflex sensitivity, and heart rate recovery after graded exercise [47].
More recently, CMR has emerged as a very useful tool for risk stratification also in patients with idiopathic dilated cardiomyopathy. Wu et al. [48]. carried out a prospective study in 65 patients with dilated cardiomyopathy with LV ejection fraction ≤35 % who underwent CMR before placement of an implantable cardioverter defibrillator for primary prevention of sudden death. Late gadolinium enhancement at CMR was found in 27 patients (42 %); during a 17-month median follow-up, 12 (44 %) of these patients had major cardiac events compared with only 3 (8 %) of those without late gadolinium enhancement. After adjustment for LV volume index and functional class, patients with late gadolinium enhancement had an 8-fold higher risk of experiencing major cardiac events.
Along the same line, Hombach et al. [49] studied a cohort of 141 patients with dilated cardiomyopathy and found that late gadolinium enhancement was detected in 36 patients (26 %). Survival analysis showed that the presence of late gadolinium enhancement, together with a QRS >110 ms, and presence of diabetes mellitus were significant predictors of a worse prognosis, although late gadolinium enhancement lost its predictive value in multivariable analysis.
Finally, Lehrke et al. [50], in a study of 184 patients with dilated cardiomyopathy who received an implantable cardioverter defibrillator, found late gadolinium enhancement in 39 % of patients. Patients with a total late gadolinium enhancement extension >4.4 % of LV mass had a 3.4-fold higher risk of the composite end point of cardiac death/implantable cardioverter defibrillator discharge/hospitalization for heart failure, as compared to those with less extensive late gadolinium enhancement (Fig. 5.8).
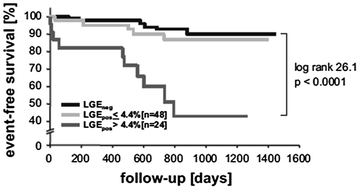
Fig. 5.8
Clinical outcome of patients with dilated cardiomyopathy according to the presence of late gadolinium enhancement (LGE), further stratified into those with an area of enhancement above or below 4.4 % of LV mass; patients with only mild enhancement had a prognosis comparable to those without evidence of enhancement. Adapted from Lehrke et al. [50]
Thus, these early data suggest that late gadolinium enhancement, reflecting the presence of fibrotic areas possibly secondary to microvascular ischemia, may have a relevant prognostic value in dilated cardiomyopathy, although this hypothesis needs confirmation in larger studies.
5.3 Aortic Stenosis
The development of LVH in patients with aortic stenosis is an adaptive response, in that it may reduce wall stress and tension in the LV. Unfortunately, hypertrophy not only provides benefits, but also has many pathological consequences, including myocardial ischemia. Angina episodes are indeed reported by about half of patients with a severe degree of aortic stenosis, and this occurs despite the evidence of normal epicardial coronary arteries at angiography [51]. Of note, the appearance of angina greatly increases the risk of sudden death, as compared to that of asymptomatic patients [52, 53].
CFR is reduced in aortic stenosis [54]. As also discussed in Chap. 2, the mechanisms of CMD in this setting are complex and include the development of severe LVH. Much of our knowledge about CBF in LVH has come from animal models. The experimental animal models, however, differ in many ways from patients with aortic stenosis, and, of course, there is no way to know whether the animal experiences angina. Nonetheless, important mechanistic data have been gleaned from these studies. First, there is a reversal of the normal subendocardial-subepicardial ratio of CBF; in normal conditions this ratio is approximately 1.2:1, appropriate to meet the physiologically higher oxygen demand of the subendocardium, where wall stress is greater than in the subepicardium. It is clear that this ratio is reduced and even reversed in LVH, especially during exercise or pacing [55–57]. Second, as noted above, CFR is impaired in LVH, due to a combination of mechanisms, including: (1) reduced time of diastolic coronary filling during exercise or pacing-induced tachycardia [55]; (2) increased LV diastolic filling pressure and intramyocardial pressure during diastole, both contributing to impairment of perfusion selectively in the subendocardium [58]; (3) reduced capillary density, with fewer capillaries per unit of myocardial mass perfused [59]; (4) a low coronary perfusion pressure as compared with intra-cavitary pressure; and (5) increased intramyocardial systolic pressure and delay in myocardial relaxation at the end of systole, which further reduces time of coronary filling and perfusion.
Post-mortem studies have confirmed capillary rarefaction in aortic stenosis while, in contrast with hypertension and HCM, they failed to find additional structural alterations which can cause CMD like medial hypertrophy or perivascular fibrosis [60].
Recent studies in patients with aortic stenosis, based on the measurement of MBF by PET, have shed new light on the mechanisms of CMD. Resting total LV MBF has been found to increase proportionally with LV mass. As this occurs in spite of vascular rarefaction, it is likely that the increase in resting MBF is mainly sustained through metabolic vasodilatation in response to the increased myocardial oxygen demand. This is responsible for a partial exhaustion of the autoregulatory capacity of the coronary microcirculation, and therefore contributes to limit CFR. These studies have also confirmed the experimental observation that subendocardial perfusion is severely reduced and have shown that the reduction of CFR is strictly related to aortic valve area and to reduced diastolic perfusion time rather than to the increase in LV mass [61] (Figs. 5.9 and 5.10). Of note, the subendocardial and subepicardial curves correlating CFR and aortic valve area intersect at 0.92 cm2, a figure that approximates closely to usually applied criteria for clinically severe aortic stenosis (Fig. 5.9).
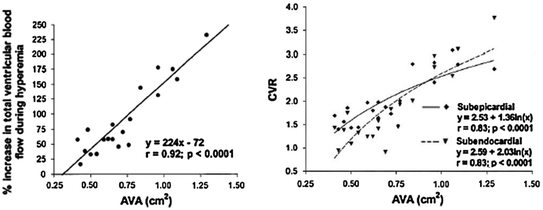
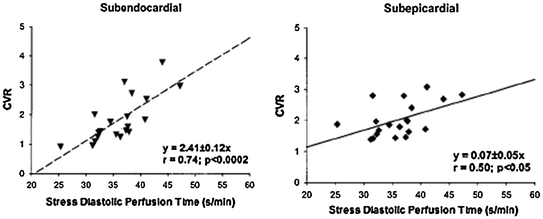
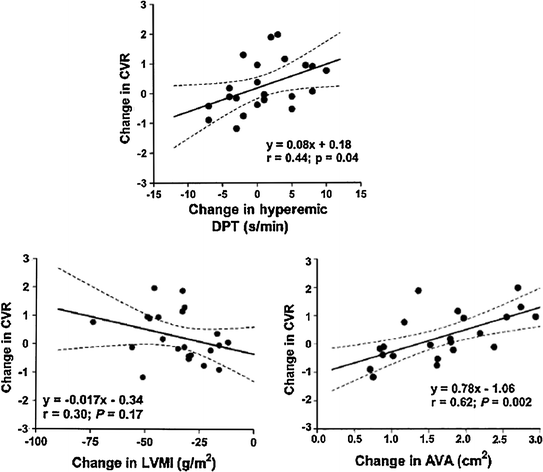
Fig. 5.9
Left: relation between aortic valve area (AVA) and hyperemic MBF in patients with aortic stenosis. Right: relation between AVA and coronary vasodilator reserve (CVR) in and CFR in subendocardial and subepicardial myocardium; progressive impairment of subendocardial perfusion is evident for decreasing values of aortic valve area. Adapted from Rajappan et al. [61]
Fig. 5.10
Relation between diastolic time and coronary vasodilator reserve (CVR) in subendocardial (left panel) and subepicardial (right panel) LV layers in patients with aortic stenosis. Reduction of diastolic time results in severe subendocardial underperfusion. Adapted from Rajappan et al. [61]
Fig. 5.11
Relation of changes in LV mass index (LVMI) (left panel), hyperemic diastolic perfusion time (DPT) (top panel), and aortic valve area (AVA) (right panel) with coronary vasodilatory reserve (CVR) in patients with aortic stenosis after aortic valve replacement. Adapted from Rajappan et al. [62]
Taken together these findings indicate that extravascular mechanisms rather than small vessel disease are responsible for the reduction of CFR in these patients. Accordingly, after aortic valve replacement the increase in CFR is directly related to the increase in aortic valve area and increase of diastolic perfusion time but not to regression of LV mass (Fig. 5.11) [62].
Interestingly, a few studies have shown that, among patients with severe aortic stenosis and normal coronary arteries, those who develop angina, as compared to those who do not develop angina, exhibit: (1) a lower CFR; (2) a higher diastolic and systolic wall stress; and (3) a reduced systolic coronary flow [63] (Figs. 5.12).
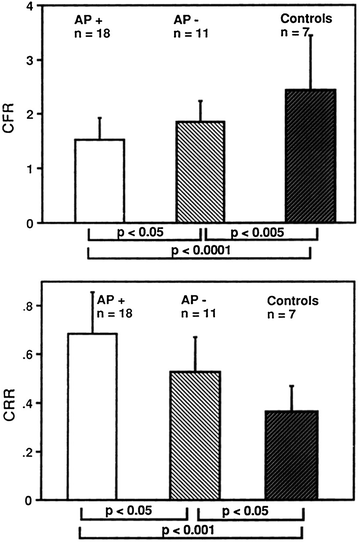
Fig. 5.12
CFR (top panel) and coronary resistance ratio (CRR) (bottom panel) in patients with aortic stenosis with anginal symptoms (AP+) or without anginal symptoms (AP−) and in control subjects. CFR was significantly reduced in AP+ patients compared with AP− patients or control subjects. Conversely, CRR was significantly increased in AP+ compared with AP- patients and control subjects. Adapted from Julius et al. [63]
The clinical implications of a significant decrease in CFR in patients with aortic stenosis are considerable. First, decreased CFR provides a plausible explanation for why ischemic symptoms such as angina pectoris are often encountered in these patients. Second, these abnormalities provide an explanation for why they often have abnormal electrocardiographic responses to exercise, suggesting myocardial ischemia even when they do not refer angina symptoms; importantly, this significantly impairs the specificity of noninvasive approaches to diagnosing atherosclerotic coronary disease including exercise ECG, exercise nuclear ventriculogram and thallium-201 perfusion scintigraphy. Third, abnormal perfusion, if severe enough, may lead to intermittent episodes of subendocardial ischemia and eventually to diffuse subendocardial fibrosis. This could in turn contribute to the development of diastolic and systolic abnormalities in ventricular function. Although there are other mechanisms that could contribute to the pathogenesis of myocardial failure in hypertrophied ventricles, it is possible that profound alterations in perfusion are the major culprit responsible for the long-term deterioration of LV function often observed in patients with aortic stenosis.
The role played by perfusion diastolic time in causing the observed reduction of CFR and angina in patients with aortic stenosis has also important therapeutic implications. Indeed, drugs that increase diastolic time, like beta-blockers, might help in delaying the ominous onset of ischemia and angina in these patients (see Chap. 9).
5.4 Myocarditis
Recent studies have shown that parvovirus B19 and human herpesvirus 6 are the most common pathogens of viral myocarditis [64] and that the clinical presentation of patients with myocarditis can be related to the type of virus present in the myocardium [65]. Patients with myocardial parvovirus B19 infection, indeed, seem to mainly present with chest pain, whereas patients with myocardial herpesvirus 6 infection or combined parvovirus B19/herpesvirus 6 infection seem to mainly show symptoms of heart failure. These findings suggest that different viruses may have specific pathophysiologic targets, although the reasons for these differences are currently unknown.
It is known, however, that endothelial cells represent specific targets in parvovirus B19-associated myocarditis [66, 67] probably through blood group P antigen [68]. Thus, a causal relationship between parvovirus B19-related myocardial inflammation and/or infection of vascular endothelial cells and CMD can be suspected [69]. In addition, myocardial inflammation in response to viral infection was found to be associated with increased endothelial expression of human leukocyte antigen system and adhesion molecules, and to be correlated with systemic endothelial dysfunction. Taken together, these findings suggest that symptoms of chest pain in patients with parvovirus B19-associated myocarditis without significant CAD may be caused by intense coronary vasoconstriction, as a result of myocarditis-induced coronary endothelial dysfunction and/or direct infection of endothelial cells and/or SMCs. Severe vasoconstriction/spasm of microcirculation might contribute to explain ST-segment elevation in some patients, which can result from sufficiently diffuse myocardial ischemia. Accordingly, in patients with parvovirus B19-associated myocarditis, Yilmaz et al. found that intracoronary acetylcholine administration was associated with severe coronary vasoconstriction [70] in distal segments of epicardial vessels, possibly extending to the coronary microcirculation (Fig. 5.13). The notion of severe CMD was confirmed in another study showing that myocardial virus persistence and myocardial inflammation were associated with endothelial dysfunction, as assessed by intracoronary administration of acetylcholine. Endothelial dysfunction in patients with myocardial virus persistence occurred independently of myocardial inflammation, although it was more pronounced in patients with concurrent inflammation [71]. Thus, endothelial dysfunction in patients with myocardial virus persistence seems to be mediated by mechanisms other than myocardial inflammatory infiltrates, which seem to also concurrently impair systemic endothelial function [72].
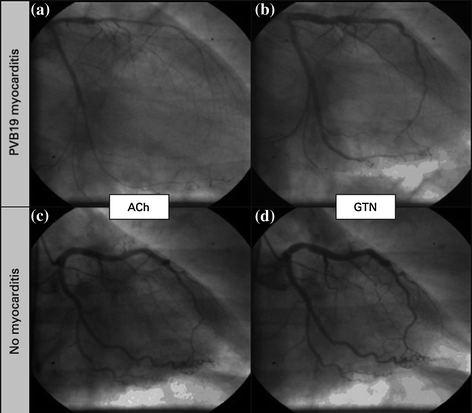
Fig. 5.13
Coronary angiograms of the left coronary artery. In a patient with parvovirus B19 (PVB19) myocarditis significant coronary vasospasm was provoked in the left anterior descending coronary artery and the left circumflex coronary artery in response to acetylcholine (ACh) (a), compared with the relaxed state after glyceryl trinitrate (GTN) (b). In contrast, no significant vasoreaction was detected in a patient with normal biopsy findings in response to ACh (c) when compared with the coronary status after GTN infusion (d). Adapted from Yilmaz et al. [70]
Parvovirus B19-associated CMD might be caused by severe endothelial dysfunction due to a decreased bioavailability of the vasodilator NO [73]. Interestingly, systemic inflammation (induced by vaccination) may cause endothelial dysfunction through reduction of vascular NO bioavailability and increases in oxidative stress [74, 75]. The hypothesis of endothelial dysfunction competes with the view of a coronary smooth muscle cell hyperreactivity as the underlying cause of coronary spasm, as assessed in experimental models [76]. In previous studies, an increased intracellular rho-kinase activity was identified as a possible key mechanism for smooth muscle cell hyperreactivity in patients with vasospastic angina [77]. Furthermore, there is evidence that the activation of this kinase may facilitate leukocyte adhesion to arteries, thereby enabling their accumulation in the vessel wall and the spread of inflammation [78]. Therefore, one might speculate that viral infection of distal coronary arteries may be associated with coronary smooth muscle cell hyperreactivity. However, it is currently unknown whether rho-kinase activity is increased in inflamed myocardium. Depending on the severity of virus infection and the individual immune response, a further spread of inflammation to other myocardial regions and layers with the accumulation of interstitial lymphocytes and macrophages can be expected, causing various patterns of myocardial damage.
In this context, increased HLA class II expression reflecting specific antigen-presenting immune cells was seen in the cardiac interstitium surrounding small-sized vessels in some patients. Hence, the infection of coronary endothelial cells with parvovirus B19 may cause a kind of ‘‘coronary vasculitis’’, which may constitute a major determinant of the future clinical course and of the myocardial spread of the inflammation.
In conclusion, parvovirus B19-associated myocarditis, with or without virus persistence, may cause severe CMD and angina. Accodingly, parvovirus B19-associated myocarditis can mimic myocardial infarction and persistence of the virus in coronary microcirculation may be responsible for persistent angina [69]. It follows that in patients who present severe rest angina, troponin raise and fall, and ischemic-like ECG changes, and are found to have angiographically normal epicardial coronary arteries, an intracoronary acetylcholine test is indicated in order to establish the presence of distal diffuse coronary vasoconstriction (different from proximal focal spasm observed in vasospastic angina). The diagnosis of myocarditis can then be established by myocardial biopsy confirming the presence of parvovirus B19. Interestingly while in myocarditis CMR typically shows mesocardial or subepicardial late gadolinium enhancement, in patients with positive acetylcholine testing a subendothelial location can be observed perhaps reflecting ischemic necrosis [70].
5.5 Infiltrative Heart Diseases
Infiltrative heart diseases are predominantly characterized by systemic alterations that cause symptoms and signs related to other organs and systems and/or by myocardial alterations responsible for symptoms and signs of heart failure. Notably, in a sizeable proportion of these patients angina is also present and, in a minority of patients, is the initial symptom. In these patients angina is typically caused by CMD; thus the occurrence of MVA, although rather infrequent, can represent the first clue to the diagnosis of a complex systemic disease.
The two infiltrative heart diseases most frequently associated with the evidence of CMD are Anderson-Fabry disease and amyloidosis.
5.5.1 Anderson-Fabry Disease
Anderson-Fabry disease is caused by an X-linked deficiency of lysosomal α-galactosidase A. It has been reported in up to 6 % of men and 12 % of women with late-onset HCM [79]. Lysosomal α-galactosidase A deficiency results in multiorgan damage determined by glycosphingolipid deposition, mainly globotriaosylceramide, with renal, cardiac and cerebrovascular involvement, and premature death [80]. In the heart, glycosphingolipid deposition is accompanied by secondary changes, such as myocyte hypertrophy, which can mimic the morphological and clinical picture of HCM as well as that of other diseases characterized by LVH (Fig. 5.14) [81]. Progressive deterioration of LV function and myocardial scarring occurs in a significant number of patients [82].