Fig. 58.1
Pathologic specimen of hypertrophic cardiomyopathy. Note the markedly thickened septum and left ventricular posterior wall (Reproduced or adapted from Driscoll, David, Fundamentals of Pediatric Cardiology, Lippincott Williams & Wilkins, 2006, with permission of the author and publisher)
The terms “obstructive and nonobstructive hypertrophic cardiomyopathy” usually implies an autosomal dominant condition that results from mutations in one of several.
58.2.2 Clinical Presentation
Patients with HC usually come to medical attention in one or more of the following ways: detection of a murmur, an abnormal electrocardiogram, from family screening or a positive family history, from evaluation of syncope, chest pain, palpitations, or out-of-hospital cardiac arrest.
58.2.3 Physical Examination
For patients with nonobstructive forms of HCM, there is no characteristic murmur. There may or may not be a prominent apical impulse and an S4. In some patients, obstruction can be unmasked and a murmur will appear with maneuvers to lower systemic vascular resistance such as inhalation of amyl nitrate or assuming a standing position.
Patients with obstructive HCM will have systolic ejection murmur. Unfortunately, in subtle cases one can mistake this murmur for an innocent flow murmur. There may be a bifid pulse.
58.2.4 Echocardiography and Cardiac Catheterization Issues
Classically there will be asymmetric septal hypertrophy and systolic anterior motion of the mitral valve. Echocardiography and Doppler will allow assessment of the absence, presence, and degree of left and right ventricular outflow tract obstruction. Echocardiography allows quantification of the thickness of the ventricular walls. Cardiac catheterization is unnecessary for diagnostic purposes but may be utilized in some adults for alcohol ablation of the obstructing septum.
58.2.5 Treatment
The most important concern is the increased risk of sudden death. Recent population-based studies indicate that the risk is 1 % per year. The ability to identify patients at high risk for sudden death is still crude, but the following features may be associated with higher risk for sudden death: diagnosis in childhood, septal thickness exceeding 30 mm, nonsustained ventricular tachycardia, failure of normal increase of systolic blood pressure during exercise, family history of premature death associated with HC, significant left ventricular outflow tract obstruction, and prior cardiac arrest.
The current paradigm for treating HC is to identify those patients at high risk for sudden death and implant an automatic internal cardiac defibrillator (AICD). For symptomatic patients who are not at high risk for sudden death and do not qualify for implantation of an AICD, treatment with a beta blocker or amiodarone can be considered.
For patients with significant left ventricular outflow tract obstruction, surgical myotomy/myectomy or alcohol ablation can be done. There is increasing evidence that elimination of the obstruction prolongs life and relieves symptoms.
58.2.6 Outcome
Untreated, there is 1 % risk of death per year. For patients with significant left ventricular outflow tract, obstruction myotomy/myectomy reduces the risk of death. For patients at high risk for sudden death (see above), implantation of an automatic defibrillator reduces the risk of sudden death.
58.3 Dilated Cardiomyopathy
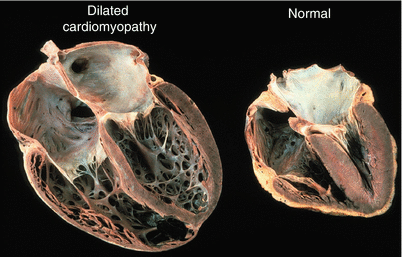
Dilated or congestive cardiomyopathy is a descriptive diagnosis implying that the left ventricle is dilated with reduced systolic function (Fig. 58.2). Normal left ventricular ejection fraction is 50–60 %. Anything less than 50 % is abnormal and could indicate the presence of dilated cardiomyopathy.