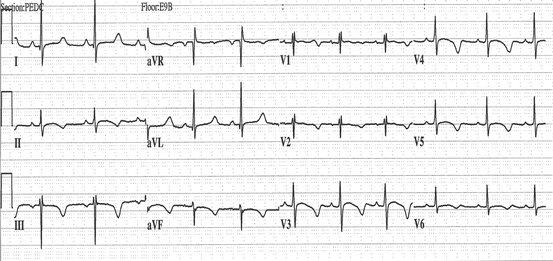
Fig. 61.1
Cardiac diagram depicting the electrical conduction system. Normal ECG
61.2 Introduction
Arrhythmias can be classified based upon the site of origin of the arrhythmia (supraventricular or ventricular), the heart rate of the arrhythmia (tachyarrhythmia or bradyarrhythmia), the function or dysfunction of the AV node (atrioventricular block or dissociation), or the underlying mechanism of the arrhythmia (reentry or bypass tract, automatic focus, ion channel defect).
Tachyarrhythmias originating from above the ventricles are termed supraventricular tachycardias and include atrial tachycardia, AV node reentry tachycardia, and tachycardia resulting from an accessory pathway (e.g., Wolff-Parkinson-White syndrome, Fig. 61.2).
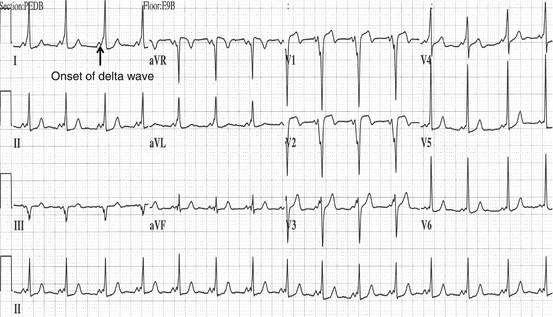
Fig. 61.2
This is an example of Wolff-Parkinson-White syndrome. Note the short PR interval and the delta wave (Reproduced or adapted from Driscoll D (2006) Fundamentals of Pediatric Cardiology. Lippincott Williams & Wilkins, with permission of the author and publisher)
Atrial fibrillation and atrial flutter, technically, are supraventricular arrhythmias but usually are simply referred to as atrial fibrillation and atrial flutter. Tachyarrhythmias originating from the ventricles are termed ventricular tachycardias.
Bradyarrhythmias (heart rate is too slow) can result from sinus node disease or AV node disease. In the latter case, the heart rate is slow because conduction through the AV node is abnormal. These bradyarrhythmias include first, second, and third degree atrioventricular block. In first degree atrioventricular block, the PR interval is prolonged but the heart rate is normal. In second degree block, some, but not all, of the atrial beats are conducted to the ventricles, and in third degree block, none of the atrial beats are conducted to the ventricle.
There are a number of arrhythmia syndromes that can be inherited, and these frequently can be associated with syncope, seizures, and sudden death. Many of these conditions result from mutations in genes encoding critical ion channels in the heart. These so-called channelopathies, a term coined by Dr. Michael Ackerman, occur in approximately 1 of 1000 persons.
61.3 Long QT Syndrome
Long QT syndrome (LQTS) is one of these (Fig. 61.3). When first described, two forms were recognized and termed Jervell and Lange-Nielsen syndrome (autosomal recessive) and Romano-Ward syndrome (autosomal dominant). Many mutations in 16 LQTS susceptibility genes have been identified. Most of these are due do to mutations in KCNQ1 causing LQT1, KCNH2 causing LQT2, and SCN5A causing LQT3. Historically, the hallmark of and diagnostic marker for LQTS was a prolonged QT interval on the electrocardiogram. However, with the advent of gene testing, it is clear that 40–50 % of patients who are genotype positive for a mutation in one of the pathologic mutations have a normal QT interval on the electrocardiogram.