Clinical Pharmacokinetics in Congestive Heart Failure
Paul Nolan
Alan S. Nies
Paul E. Fenster
Pharmacokinetics is the study of drug movement in the body from the time of dosing to the time at which all of the drug has been eliminated. Knowledge of pharmacoki-netics is key to the design of dosing regimens for optimizing a drug’s therapeutic effect. Drug movement across short distances, such as from the lumen of the gastrointestinal tract into the intestinal mucosa or across a cell membrane in the body, is not dependent on blood flow. Intuitively, the circulation must be an important determinant of a drug’s movement over longer distances. However, only in the past two decades has there been an attempt to model and quantify the effect of changes in circulatory function on various pharmacokinetic parameters (1,2,3,4). It is now known that all aspects of pharmacokinetics (i.e., absorption, distribution, and elimination) are critically dependent on the circulation. Changes produced by heart failure that would be expected to influence these pharmacokinetic parameters include (a) reduced blood flow to sites of drug absorption such as skin, subcutaneous tissue, or intestine; (b) interstitial edema at sites of absorption in the intestine or skin; (c) delayed gastric emptying due to increased sympathetic and reduced parasympathetic tone; (d) reduced blood flow to tissues that normally store drug in the body, such as fat and muscle; (e) reduced blood flow to the liver; (f) hepatocellular damage due to hypoxia or congestion; and (g) reduced blood flow to the kidney with resulting changes in renal function (5,6,7). Data exist for some model compounds regarding the influence of an altered circulation on phar-macokinetic variables, and from these data certain generalizations can be made to guide therapy with drugs for which data do not yet exist. This chapter starts with the basics. A description of the important aspects of pharma-cokinetics as they relate to the circulation is followed by information on specific drugs that are important for the treatment of patients with congestive heart failure (CHF).
Pharmacokinetic Principles
Absorption
To produce systemic effects, drugs must be delivered into the central circulation (Table 23-1). When a drug is delivered directly into the vascular space, all of the drug administered is available to produce its effect. For other routes of administration, the drug must pass a variety of barriers before it is able to gain access to the circulation. This absorption process will delay delivery of drug to its site of action and in some circumstances will reduce the total quantity of drug reaching the circulation. At a minimum, a single cell layer, the capillary endothelium, must be traversed even when a drug is administered intramuscularly. Much more complex is absorption after oral administration where several cell layers must be crossed before the drug reaches the portal circulation, after which the drug must make it through the liver before it reaches the systemic circulation. Many β-adrenergic receptor antagonists, calcium-channel blockers, antiarrhythmic agents, and nitrates are substantially metabolized by the liver before they reach the systemic circulation. This presystemic or first-pass elimination accounts for the much larger oral dose required to produce the same effect as an intravenous dose. Bioavailability is the term that quantifies the amount of a dose that actually reaches the systemic circulation and varies on a scale from O (none of the drug reaches the circulation) to 1 (100% of the dose reaches the systemic
circulation). A reduction of bioavailability after oral administration of a drug indicates either poor absorption or presystemic elimination (8).
circulation). A reduction of bioavailability after oral administration of a drug indicates either poor absorption or presystemic elimination (8).
Table 23-1 Influence of Heart Failure on Pharmacokinetic Factors Influencing the Plasma Concentration of Drugs | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||
Most drugs are absorbed from their site of administration by passive diffusion, with the driving force for absorption being the concentration of drug in contact with the absorbing surface. Thus, the rate of absorption can be manipulated by controlling the dissolution of drug from the dosage form. This principle has been useful for designing drug preparations that delay and prolong absorption after oral, intramuscular, subcutaneous, or percutaneous sites of administration. In this way drugs that have short durations of action can be used for chronic therapy with relatively infrequent dosing. Depot preparations for intramuscular use (e.g., penicillin and progesterone) slowly release drug, which then is dissolved in tissue fluids and absorbed into the circulation, sustaining an effect over days or weeks. Long-acting oral preparations (e.g., calcium-channel blockers, procainamide, and theophylline) release drug slowly from the dosage form. For these oral preparations, the duration of sustained absorption is limited to 12 to 24 hours by the transit time of the gastrointestinal tract. Percutaneous absorption is achieving increasing use for a few, highly potent, lipid-soluble drugs. The duration of absorption is limited by the ability to incorporate sufficient drug into the patch that is applied to the skin. The absorption of percutaneous clonidine is sustained over a week; nitroglycerin is administered daily. The advantages of these
sustained-release preparations are that they are more convenient and thus have the ability to enhance compliance, and they produce a relatively constant blood level so that toxicity associated with the peaks and inefficacy associated with troughs of blood concentration are avoided.
sustained-release preparations are that they are more convenient and thus have the ability to enhance compliance, and they produce a relatively constant blood level so that toxicity associated with the peaks and inefficacy associated with troughs of blood concentration are avoided.
With preparations other than oral, drug can reach the systemic circulation without having to pass through the liver and be subjected to hepatic presystemic metabolism. This can improve the bioavailability for a drug, such as nitroglycerin, which is essentially metabolized completely by the liver after oral administration. However, presystemic metabolism may not be avoided entirely because skin also may metabolize nitroglycerin before it reaches the circulation during percutaneous absorption. A disadvantage is that interindividual variation in absorption is increased with the sustained-release preparations; with oral dosage forms, sustained-release preparations are more likely to be affected by changes in gastrointestinal motility such that increases in motility can reduce the total quantity of drug absorbed. Another disadvantage that is unrelated to pharmacokinetics is the increased potential for the development of tolerance to the drug’s effect when blood levels are sustained relatively constant over long periods of time. This has been of particular interest with the use of percutaneous nitroglycerin, which is discussed elsewhere in this book.
Factors other than drug concentration are important for absorption, particularly in disease states such as CHF (5). Resistance of the absorbing surface to diffusion is a function of the area available for absorption as well its permeability to the drug. Blood flow to the absorption site also is important to remove drug and thereby maximize the concentration gradient across the absorbing surface (9). For drugs that dissolve readily from the dosage form and diffuse rapidly across cells, blood flow to the site of absorption may be rate-limiting. For most drugs, however, the rate of drug delivery from the dosage preparation or the resistance of the absorbing surface to diffusion is the ratelimiting step in absorption such that alterations in blood flow become less important. In CHF, edema and a reduction of blood flow at the absorbing site have the potential to reduce bioavailability and/or alter the time course of drug absorption (10). After oral dosing, most drugs are absorbed across the mucosa of the small intestine so that a delay in gastric emptying can retard absorption. This most commonly delays and decreases the height of the peak concentration in the blood. In heart failure, the sympathetic nervous system is activated and the parasympathetic nervous system is depressed. In the gastrointestinal tract these consequences of heart failure reduce peristaltic activity and delay gastric emptying. Thus, several features of heart failure, including edema formation, a reduction of blood flow, and a delay in gastric emptying, would be predicted to reduce the rate or extent (i.e., the bioavailability) of absorption of some drugs (11). It is important to recognize the consequences of a reduction in the rate or extent of drug absorption. For drugs such as digoxin and theophylline that are administered chronically and for which the target blood levels must remain within a certain range for therapeutic effect, a delay in the rate without a decrease in the extent of absorption is of little or no importance. In fact, a delay in absorption will result in a more constant blood concentration than would be achieved if the dose were absorbed rapidly. However, the rate of absorption can be important if the circulating drug concentration must reach a threshold level or if the rate of drug delivery to the site of action is a critical determinant of the drug’s effect. This is the case for furosemide, which has a renal response that is determined by the rate as well as the extent of drug reaching the tubular fluid (12). A reduction in the rate of oral absorption is one mechanism accounting for the resistance to the diuretic effect of oral furosemide in patients with heart failure.
In contrast to the relative unimportance of a reduction in rate of absorption for most drugs, a reduction in extent of absorption (bioavailability) will be of importance for all drugs affected because the steady-state concentration of the drug in the plasma will be reduced. The limited amount of data available in CHF indicates that the extent of drug absorption by the gastrointestinal tract usually is affected only minimally and unpredictably. However, the rate of drug absorption from the gut often is delayed, probably as a consequence of a delay in gastric emptying, although edema of the gut wall and a diminished blood flow also could play a role. With transdermal nitroglycerin, there is some evidence that bioavailability is variably reduced in patients with severe CHF, probably as a consequence of subcutaneous edema and reduced dermal blood flow.
Distribution
Once absorbed into the blood, drugs are distributed throughout the body, initially to the well-perfused tissues, including the vasculature, heart, and brain, and then to the less well-perfused tissues, including fat and skeletal muscle. The extent of uptake into tissues depends on the ability of the tissues to bind or partition the drug relative to the binding or partitioning in the plasma. The pharmacoki: netic term that describes the distribution of drugs is called the apparent volume of distribution (VD). VD is defined as the volume that would be required to contain an amount of drug at the concentration achieved in plasma, and therefore is a constant that describes the ratio of the amount of drug in the body (Amt) to the resulting plasma concentration (CP):
Depending on the rate of drug entry into the circulation, the body often behaves as if it were composed of two or more compartments that each have their own VD. Drug is introduced initially to a smaller VD (often designated as the central compartment) and only more gradually is distributed to a larger VD (often called the peripheral compartment). Thus, immediately after an intravenous bolus of a drug such as lidocaine, the drug concentration in the blood is high, but the drug rapidly disappears from the central compartment as it is distributed to the peripheral compartment. The drug concentration then decreases more gradually as drug is eliminated from the body.
The two phases have been called the initial distribution phase followed by the elimination phase. The most important phase for chronic drug therapy is the elimination phase, during which drug is in equilibrium between the
blood and the tissues of the body, and the concentration of drug in the blood therefore bears a consistent relationship to drug at the site of action. Therefore, the VD describing this apparent volume of the peripheral compartment is the most clinically relevant value for most drugs and is the VD listed in tables describing drug pharmacokinetics. Whether the drug concentration in plasma during the distribution phase is predictive of drug effects is dependent on the individual drug. For many drugs such as lidocaine that quickly reach their site of action, plasma concentrations achieved during the distribution phase are associated with therapeutic or toxic effects on the well-perfused tissues such as the heart and brain. On the other hand, for a few drugs like digoxin, the plasma concentrations during the distribution phase are not predictive of drug effects. Only after 4 to 8 hours is there sufficient time for equilibrium to occur such that digoxin in the blood reflects drug at the site of action. A common error in the interpretation of digoxin concentrations in plasma is to ascribe meaning to the high values obtained when blood is sampled within 4 to 6 hours after a dose of digoxin (13). Although concentrations of 4 to 10 ng/mL may be present at this time, they have no predictive value because they are not in equilibrium with digoxin at its site of action.
blood and the tissues of the body, and the concentration of drug in the blood therefore bears a consistent relationship to drug at the site of action. Therefore, the VD describing this apparent volume of the peripheral compartment is the most clinically relevant value for most drugs and is the VD listed in tables describing drug pharmacokinetics. Whether the drug concentration in plasma during the distribution phase is predictive of drug effects is dependent on the individual drug. For many drugs such as lidocaine that quickly reach their site of action, plasma concentrations achieved during the distribution phase are associated with therapeutic or toxic effects on the well-perfused tissues such as the heart and brain. On the other hand, for a few drugs like digoxin, the plasma concentrations during the distribution phase are not predictive of drug effects. Only after 4 to 8 hours is there sufficient time for equilibrium to occur such that digoxin in the blood reflects drug at the site of action. A common error in the interpretation of digoxin concentrations in plasma is to ascribe meaning to the high values obtained when blood is sampled within 4 to 6 hours after a dose of digoxin (13). Although concentrations of 4 to 10 ng/mL may be present at this time, they have no predictive value because they are not in equilibrium with digoxin at its site of action.
The VD is an apparent volume that is rarely related to an actual volume, and it is a common mistake to attempt to label the VD as plasma volume, extracellular water, or some other actual fluid space. For drugs that are bound to tissues, the VD can be much larger than any real volume contained in any fluid space in the body. For instance, digoxin has a VD of the peripheral compartment of 7 L/kg in normal individuals. Thus, an intravenous dose of 0.5 mg digoxin will be distributed in a peripheral compartment that has an apparent volume of about 500 L in a 70-kg patient, resulting in a plasma concentration of 1 μg/L or I ng/mL. A large VD is the quantitative expression of the fact that drug in the body is present in the tissues at a higher concentration than in the blood. Even drugs that have a VD similar to a body fluid compartment are rarely distributed everywhere at the same concentration.
The physiological determinants of the VD are the relative affinities of the tissues and the plasma for the drug and the state of the circulation (2,5,14). Binding of drug to plasma proteins acts to retain drug in the plasma because it limits the amount of unbound drug that can distribute to the tissues. Displacement of drug from plasma proteins will increase its VD because there is an increase in the fraction of drug that is unbound, which will leave the central compartment. Drug binding to plasma proteins can be affected by diseases and other drugs. Uremia can reduce the binding of some drugs to plasma albumin because retained acidic metabolites can compete for binding sites. Nephrotic syndrome and hepatic disease can reduce the albumin concentration and thereby reduce the binding of drugs to plasma proteins. CHF usually is not associated with changes in binding to plasma proteins unless it is also accompanied by renal or hepatic failure. However, the state of the circulation itself is apparently an important determinant of the VD. This is probably related to the fact that drug is not as readily delivered to the tissues when the circulation is impaired (15). During heart failure, intense peripheral vasoconstriction restricts access of drug to poorly perfused tissues that make up the peripheral compartment of the VD. Thus, drugs that are extensively distributed out of the plasma, and therefore have a large VD, may have a contraction of the VD in patients with heart failure. This is perhaps counterintuitive on first glance because the edema present in patients with heart failure might suggest that the VD should be increased. However, this is a fallacy based on thinking of the VD as a real volume. One model compound for which the VD has been shown to be increased is aminopyrine, a substance that actually does seem to distribute only in body water (16). For most drugs the following rule generally holds: if the VD is large (1 l/kg or more) then the VD may be reduced in patients with heart failure; for drugs with small VD (<1 L/kg), there is little or no change in VD with heart failure. Lidocaine is the best-studied example of a drug that has a reduced VD of the central and peripheral compartments by about 50% in patients with heart failure (17).
The size of a loading dose (LD) is determined by the volume of distribution of the drug. The concept is that sufficient drug must be given such that after the distribution phase, the plasma concentration is in the desired range (Cdesired). Generally, the VD of the peripheral compartment is the constant used to calculate an LD delivered into the circulation by rearranging Equation 23–1:
For many drugs the calculated LD must be administered slowly to allow time for distribution so that the very high concentrations resulting from the initial distribution into the small VD of the central compartment are avoided. In the presence of heart failure, LDs of some drugs must be reduced to account for the reduced VD in this condition. For lidocaine, therefore, the loading boluses in a patient with heart failure should be about half the standard doses because the VD for lidocaine is reduced by half in such patients.
Elimination
Most drugs are eliminated from the body by the liver, the kidney, or both organs. For a few drugs, elimination occurs in the lung, the blood, or in other tissues. For most drugs, the elimination rates are directly proportional to the concentration of drug in the plasma, a process mathematically described as first-order elimination. The most useful concept for describing the variables that are physiologically important for drug elimination is drug clearance. Drug clearance usually is defined as the volume of fluid (blood or plasma) completely cleared of drug in a unit of time, so clearance has units of flow. Clearance was a concept originally used by renal physiologists to describe renal function. Creatinine clearance is the volume of plasma that is completely cleared of creatinine per minute and is calculated as the urinary elimination rate of creatinine divided by the plasma creatinine concentration. By analogy, drug clearance (Cl) can be considered as the rate of drug elimination (Re) normalized to the plasma concentration (Cp) (4):
The total body drug clearance is the sum of all the individual organ clearances (renal clearance plus hepatic clearance plus lung clearance plus clearance by alt other means).
Physiologically, clearance is equal to the product of blood flow (Q) to the eliminating organ and the extraction ratio (E), which is the fraction of the drug in the blood removed during a single passage through the organ:
Clearance is thus an index of the efficiency of drug removal from the blood and is not influenced by the distribution of drug in the body. Equation 23-4 implies that hepatic drug clearance (ClH) would be related directly to hepatic blood flow. However, this is not the case because the extraction ratio is not a constant. Hepatic extraction of drug from the blood is a function of (a) hepatic blood flow, (b) the inherent ability of the liver to irreversibly remove drug from the blood, and (c) the binding of drug to the plasma proteins and the cellular components of blood (2,3). A number of models of hepatic elimination have been proposed (4). The most useful is the well-stirred model, which assumes that drug available to the eliminating processes of the liver is in equilibrium with hepatic venous blood (1). Based on this model the following relationship can be derived:
where fB is the free (unbound) fraction of drug in the blood and Cluint is the intrinsic hepatic clearance, an index of the ability of the liver to remove unbound drug from liver water by metabolizing enzymes and transport processes. Because binding of drug to plasma proteins is not changed by heart failure, fBCIuint can be combined into a single term, CIint, which represents the ability of the liver to remove drug from blood rather than from liver water. Although Equation 23-5 appears formidable, it is useful for predicting the changes in hepatic drug clearance that occur when hepatic blood flow is impaired in CHF. It is instructive to consider two drug types that represent the limits of this equation. First consider a drug with a very high intrinsic hepatic clearance. In this case, CIint is much larger than hepatic blood flow, and so the extraction ratio [CIint/(Q + CIint)] is very high and approaches 1. Thus, hepatic drug clearance (Eq. 23-5) approaches hepatic blood flow. Drugs that have this type of CIint are called high-clearance drugs and include lidocaine, propranolol, nifedipine, diltiazem, and verapamil. When given intravenously, their clearance is dependent on hepatic blood flow. Because of the very high hepatic extraction, such drugs are subject to a high first-pass hepatic extraction and consequently have poor oral bioavailability. Contrast this with the situation when CIint is much less than hepatic blood flow. In this case the extraction ratio approaches [CIint/Q], and hepatic drug clearance therefore approaches CIint and is independent of hepatic blood flow. Such drugs are called low-clearance drugs and include warfarin, theophylline, quinidine, and mexiletine. Thus, the model predicts that the hepatic clearance of drugs with a high hepatic extraction ratio and, hence, a high hepatic clearance are very sensitive to changes in hepatic blood flow, whereas the clearance of drugs with a low extraction ratio and consequently a low hepatic clearance are insensitive to changes in blood flow. This is because the extraction of these low-clearance drugs is inversely proportional to hepatic blood flow so that as blood flow is reduced, the extraction ratio increases nearly proportionally, and clearance, which is the product of blood flow and extraction, is therefore little changed. This is illustrated graphically in Figure 23-1. Figure 23-1A illustrates the hepatic clearance (solid line) and hepatic extraction ratio (dashed line) versus hepatic blood flow for a high-clearance drug that has a hepatic extraction of 0.9 at a normal liver blood flow of 1,500 mL per minute. Clint is assumed to remain constant as hepatic blood flow changes. As hepatic blood flow varies from 400 mL per minute to 2,000 mL per minute, the actual hepatic clearance for the high-clearance drug varies almost proportionally and the extraction ratio remains relatively constant. In Figure 23-1B, the effect of changes in hepatic blood flow on the actual hepatic clearance (solid line) and extraction ratio (dashed line) is depicted for a low-clearance drug that has a hepatic extraction ratio of 0.1 at a normal liver blood flow of 1,500 mL per minute. Here the hepatic clearance remains relatively constant as liver
blood flow changes because the extraction of the low-clearance drug is increased as hepatic blood flow is reduced (2,3).
blood flow changes because the extraction of the low-clearance drug is increased as hepatic blood flow is reduced (2,3).
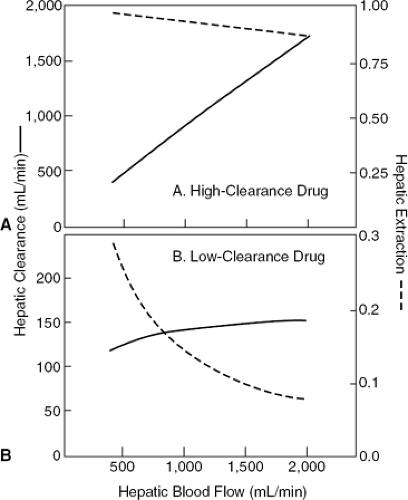 Figure 23-1 The effect of changes in hepatic blood flow on the actual hepatic clearance (solid line) and extraction ratio (dashed line) of two drugs that are metabolized entirely by the liver. A high-clearance drug (E = 0.9 at a liver blood flow of 1,500 ml per minute) is shown (A) and a low-clearance drug (E = 0.1 at a liver blood flow of 0.1 ml per minute) is shown (B). Liver blood flow is varied over the range of 400 to 2,000 mL per minute. (Modified from Wilkinson GR, Shand DG. A physiological approach to hepatic drug clearance. Clin Pharmacol Ther. 1975:18:377–390 .) |
A similar series of arguments can be applied to changes in hepatic function related to high- and low-clearance drugs. In the model, changes in hepatic function are reflected by changes in Clint. Thus, the hepatic clearance of low-clearance drugs is influenced primarily by changes in hepatic function and is relatively independent of changes in blood flow, and vice versa for high-clearance drugs. Severe CHF can produce hepatocellular damage from hypoperfusion or congestion, which will result in a reduction of Clint. Therefore, because heart failure can reduce hepatic function as well as hepatic blood flow, the hepatic clearance of both high-clearance and low-clearance drugs can be affected, but for different reasons.
A powerful prediction from the model that is not intuitively obvious is that the average plasma concentration of an orally administered drug that is eliminated solely by hepatic metabolism is not affected by changes in hepatic blood flow, even if the drug is a high-clearance drug. This is because a change in hepatic blood flow will alter the extraction of drug not only on the first pass through the liver but also on each subsequent pass, and these influences cancel each other. As shown in Figure 23-2, a reduction in hepatic blood flow will increase the first-pass hepatic extraction (E) of a high-clearance drug (Eq. 23-5), reducing the amount of an oral dose reaching the systemic circulation and thereby reducing the peak plasma concentration achieved after the dose. However, this reduced amount of drug is then cleared less well because of the decrease in hepatic blood flow (Eq. 23-5), which, if VD is unchanged, will prolong the drug’s half-life. This reduction in hepatic clearance exactly counters the reduced amount escaping hepatic extraction on the first pass, resulting in no change in average plasma concentration. Mathematically, the apparent clearance of a hepatically metabolized drug administered orally (ClO) is the hepatic clearance (ClH) divided by the fraction of the dose escaping first-pass hepatic extraction (1 – E). By substituting in Equation 23-5 and solving for ClO, one derives the surprising fact that:
indicating that the apparent clearance for a drug given orally is independent of hepatic blood flow and only dependent on the Clint, which is the functional capacity of the liver to remove drug from blood. Clinically, Equation 23-6 suggests that if heart failure produces changes in steady-state blood levels for any given oral dosing regimen of a drug that is solely eliminated by hepatic metabolism, then there must be a reduction in hepatocellular function. It is only for intravenous infusions of high-clearance drugs, such as lidocaine, that hepatic blood flow has a significant effect on steady-state blood levels.
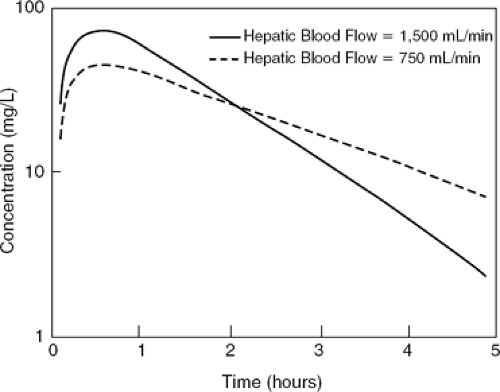 Figure 23-2 The effect of a change in hepatic blood flow on the plasma concentration-time profile of a high-clearance drug (E = 0.9 at a liver blood flow of 1,500 mL per minute) that is metabolized entirely by the liver and is administered orally at time 0. The curve with a normal liver blood flow is depicted by the solid line, and the curve with a liver blood flow of half normal (750 mL per minute) is depicted by the dashed line. When liver blood flow is reduced, the peak plasma concentration is reduced, indicating an increased hepatic presystemic elimination. However, once in the circulation, the drug is eliminated less rapidly because the hepatic blood flow is reduced. As a result of these two effects of a reduction in hepatic blood flow, the average plasma concentration (i.e., the area under the curve) is unchanged. (Modified from Wilkinson GR, Shand DG. A physiological approach to hepatic drug clearance. Clin Pharmacol Ther. 1975;18:377–390 .) |
Renal clearance of drugs is much simpler and more predictable than hepatic drug clearance. Fortunately, drug clearance by the kidney is proportional to glomerular filtration rate even if the drug is cleared by tubular secretion and/or reabsorption rather than by filtration. Thus, by estimating or measuring creatinine clearance, one can determine the effect of renal dysfunction on renal drug clearance, and dosage adjustments can be made accordingly (8).
Half-Life
The most commonly used pharmacokinetic term is drug half-life (t1/2), which is the time required to reduce the plasma concentration of drug by half. Half-life often is used to characterize drug elimination. However, it frequently is not appreciated that half-life is actually a hybrid term that is dependent on two independent variables, drug clearance (Cl) and VD:
Thus, at any given clearance, more time will be required (i.e., a longer half-life) to clear the body of drug if there is a large VD than if the VD is smaller. If clearance is reduced, half-life will be prolonged as long as VD is not changed. Thus, a change in half-life cannot be used as an index of efficiency of drug elimination unless the apparent volume of distribution is unchanged. Cardiac failure can reduce the clearance as well as the volume of distribution of some drugs; therefore, half-life is a particularly poor index of changes in drug elimination in this disease state. The best example is that of lidocaine, which has a reduction of both clearance and VD of about 50% in some patients with heart failure. However, because of the relationship in Equation 23-7, half-life may not be changed. Nonetheless, the loading doses and maintenance doses of lidocaine must be reduced to avoid toxicity. If only half-life is measured,
there will be no clue to the major changes in kinetics that have occurred in this illness.
there will be no clue to the major changes in kinetics that have occurred in this illness.
Half-life is important for the time course of drug elimination and accumulation. If drug administration ceases, only half the drug leaves the body in one half-life, half the remaining drug is eliminated in the second half life, and so forth. Thus, if 100% is present at time 0, 50% will be present after one half-life, 25% after two half-lives, 12.5% after three half-lives, 6.25% after four half-lives, and 3.125% after five half-lives. In theory, an infinite time is needed to rid the body of all drug; in practice, three to five half-lives are needed to effectively eliminate drug.
When drugs are given as a continuous infusion or as repeated intermittent doses, drug accumulates in the body until a steady state is achieved. The rate of drug accumulation depends on drug half-life and is the mirror image of the rate of drug elimination previously discussed. Thus, accumulation to half of the ultimate steady-state level occurs in one half-life, but it takes three half-lives to achieve 87.5% of steady state and five half-lives to reach 96.875% of steady state. For practical purposes, steady state is achieved when 90% of the ultimate accumulation occurs. For drugs with a long half-life accumulation occurs slowly, and an LD may be necessary to achieve therapeutic effects quickly. However, regardless of whether a loading dose is given, the final steady state achieved is independent of the loading dose or the half-life and is determined solely by the maintenance dose and the drug clearance (discussed later). The concept of accumulation applies equally well to a continuous infusion or to intermittent doses. Because smaller doses are given more frequently, the variation in peaks and troughs diminishes, but the average blood level reached at steady state is independent of dose frequency and is determined only by the dose per unit time and the clearance.
Half-life also describes the time course of changes in drug concentration when the dose or infusion rate is changed. Using the same reasoning as previously discussed, three to five half-lives will be required to achieve a new steady state. Thus, the consequences of changing doses or infusion rates are delayed, and this delay is dependent on the half-life.
Maintenance Doses
When the amount of drug administered and absorbed over a period of time (dose/time, D/t) is the same as the amount of drug eliminated during the same time (elimination rate, Re), steady state has been achieved. From the definition of clearance in Equation 23-3, it can be readily appreciated that both the dose/time and the elimination rate are equal to drug clearance multiplied by the steady-state plasma concentration (Css):
Thus, drug clearance and the dose/time are the sole determinants of the concentration of drug in the blood and the amount of drug in the body at steady state. Importantly, the steady-state concentration is independent of the VD or the half-life. For drugs given intra-venously, hepatic clearance is dependent on both hepatic blood flow and hepatic function, which therefore influence the steady-state concentration. For drugs given orally, the hepatic portion of drug clearance is independent of hepatic blood flow and only dependent on the liver’s metabolic capacity (Eq. 23-5). Equation 23-8 can be used to calculate the dose required to achieve a given plasma concentration of the drug if clearance is known. As an example, the clearance of lidocaine in an average adult without heart failure or hepatic disease is about 800 mL per minute. Thus, achieving a therapeutic plasma concentration of 3 mg/L will require an infusion of 2.4 mg per minute. In a patient with heart failure and reduced hepatic blood flow, lidocaine clearance may be reduced to 400 mL per minute, and thus an infusion rate of 1.2 mg per minute will be all that is necessary to achieve the same therapeutic level of 3 mg/L.
Effect of Heart Failure on Pharmacokinetics of Specific Drugs
Antiarrhythmic Drugs
Quinidine
Much of the early data on quinidine kinetics in heart failure are confusing because of nonspecific assay methodology (9,18). Oral absorption can be delayed in some patients with heart failure, and the VD (normally 2.7 L/kg) may be reduced such that quinidine concentrations peak later and are higher after an oral dose in patients with heart failure (19,20). Quinidine is largely metabolized by the liver as a low-clearance drug (normally 4.7 mL/min/kg). However, if heart failure is severe enough to cause hepatocellular dysfunction, the clearance of quinidine will be reduced so that higher blood levels will result from usual oral doses (21,22). Quinidine’s half-life (normally 6 to 7 hours), however, is not changed in most patients with heart failure because of an equivalent reduction in VD and clearance. However, an occasional patient with severe heart failure and hepatic dysfunction with markedly depressed hepatic quinidine clearance will have a prolonged half-life (23).
Quinidine has pharmacokinetic drug interactions with a number of drugs. Its metabolism can be induced by several common enzyme inducers, including phenobar-bital (24), phenytoin (24), and rifampin (25), resulting in an increase in hepatic clearance as a consequence of an increase in Clint. Quinidine plasma concentration therefore will decrease as a result of this interaction and may fall below the therapeutic range. Cimetidine (26), verapamil (27), and amiodarone (28) can reduce the metabolism of quinidine and thereby reduce its clearance, resulting in an increase in steady-state quinidine levels. Quinidine and verapamil also produce additive effects on the vasculature, and the combination can result in severe hypotension (29). Importantly, quinidine can reduce the clearance of digoxin (30) and, to a lesser extent, digitoxin (31), increasing the blood levels of these glycosides. Finally, quinidine can block the enzyme that accounts for the rapid metabolism of encainide and propafenone in 93% of the population who are extensive metabolizers of these drugs (32,33).
The consequences of this interaction are complex because both encainide and propafenone have active metabolites, and whether dosage adjustments are required is not clear.
The consequences of this interaction are complex because both encainide and propafenone have active metabolites, and whether dosage adjustments are required is not clear.
Procainamide
Procainamide is normally well-absorbed from the gastrointestinal tract, but in the presence of severe heart failure absorption may be delayed and/or incomplete. The VD of procainamide (normally 1.9 L/kg) has been found to be reduced in some patients with heart failure (34), but this has not been a universal finding (35). Procainamide is metabolized to an active metabolite, N-acetylpro-cainamide (NAPA, acecainide), which has class III antiar-rhythmic activity. The metabolism rate is genetically determined, with about half of the American population being fast and half slow acetylators. In all patients with normal renal function, 60% to 80% of procainamide and essentially all of the acetylated metabolite is excreted in the urine. Hepatic clearance of procainamide is low and not affected much by liver blood flow. The major determinant of the drug’s clearance, therefore, is the renal function (36). When renal function is diminished, the acetylated metabolite accumulates more than the parent drug, and this can be the case in heart failure-induced renal dysfunctions (37). Thus, for optimum patient management, the blood concentrations of both procainamide and acecainide should be monitored. In the absence of renal dysfunction, patients with heart failure do not have any consistent changes in procainamide clearance and therefore require drug doses similar to patients without heart failure to maintain therapeutic concentrations of the drug in the blood (35).
Disopyramide
Disopyramide is contraindicated in patients with heart failure because of its potent negative inotropic effects (39,40,41). Therefore, there are not many studies of its kinetics in this situation. Oral absorption of disopyramide is not changed by heart failure. The clearance of disopyramide (normally 1.2 mL/min/kg) is about 55% renal, with the remainder being hepatic metabolism. Clearance can be reduced in heart failure, and half-life (normally 6 hours) can be prolonged, especially if there is renal dysfunction (42). The VD of disopyramide is relatively small (normally 0.6 L/kg) and is unchanged in heart failure.
The most important interactions with disopyramide are with other negatively inotropic drugs such as β-adrenergic receptor antagonists that can add to the cardiac depression produced by disopyramide. A few pharmacokinetic interactions with potential clinical importance have been reported, including a reduction of disopyramide clearance with hepatic P450-inducing drugs (43), such as phenobarbital, phenytoin, or rifampin, and inhibition of disopyramide metabolism with erythromycin (44).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


