Fig. 25.1
Familial Cardiomyopathies and genetic substrate (Image modified from original image courtesy of EKKAN)
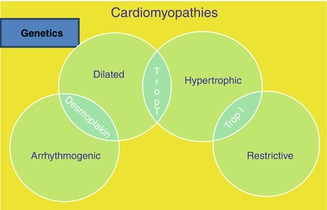
Fig. 25.2
Cardiomyopathies and overlapping in genetic substrates (Image reproduced courtesy of EKKAN)
25.1.1 Incomplete Penetrance and Variable Expressivity
Inherited CMPs show marked phenotypic variability, even within families [1–4]. Penetrance, the proportion of mutation carriers with clinically detectable disease increases with age but remains less than 100 %. In most persons with hypertrophic CMP, hypertrophy is manifested in adolescence, whereas the age at onset in patients with sarcomeric dilated CMP is bimodal, with a peak during childhood and mid-adulthood [5]. The disease is gradually progressive in patients with dilated CMP due to laminopathy (LMNA) [1, 2]. It is uncommon to find numerous persons with clinically apparent arrhythmogenic right ventricular cardiomyopathy (ARVC) in a single pedigree, which indicates a low level of penetrance [1, 2]. Early reports of all CMPs described, as expected, patients with severe forms of the disease. Subsequent studies, however, have shown that most of the affected persons have mild, sometimes atypical forms of the disease; as a result, the number of cases in a given family, and thus the proportion of familial cases, is greater than originally suspected. Only a minority of patients with hypertrophic CMP have the classic feature of outflow obstruction at rest, and up to half the cases of idiopathic dilated CMP are familial [1–8]. It has also become apparent that ARVC often goes unrecognized and is more common than was first thought [1, 2]. Incomplete penetrance requires diagnostic criteria of less than the usual stringency for first-degree relatives; clinicians caring for families at risk now use complex diagnostic algorithms to interpret minor abnormalities [1, 2]. The corollary is that, in the general population, patients with subtle features of inherited CMP are difficult to recognize. Thus, population screening is generally ineffective; instead, cascade screening (sequential identification of related family members, increasingly guided by genetic testing) is the key.
25.1.2 Genetic Heterogeneity and Genotype–Phenotype Correlations
Hypertrophic and dilated CMP can be allelic, each caused by specific missense mutations in the same genes encoding sarcomeric proteins [2]. Since these diseases arise from mutations with opposing biophysical consequences [2], a variant “breeds true” within each family; there has been no reliable documentation of families in which a single sarcomere mutation causes hypertrophic CMP in some members and dilated CMP in others. However, other aspects of the CMP phenotype can vary within families, indicating the absence of a precise relationship between the mutation and its biophysical consequences [1, 2]. For example, familial restrictive CMP is part of the spectrum of sarcomeric hypertrophic CMP, with a loose relationship between certain mutations and this variant of the phenotype [1, 2, 5, 8].
In certain circumstances, knowledge of the gene underlying the CMP may alter patient care. One example is forms of hypertrophic CMP with different inheritance patterns and natural histories [8–12]. Another example is the susceptibility to disturbances of the conduction system of patients with dilated CMP due to LMNA mutations; when this is sufficient to warrant pacemaker insertion, use of an implantable cardioverter – defibrillator should be considered [1, 2]. However, for most CMPs, correlation between the disease gene and the phenotype are currently of limited utility for managing the care of individual patients [4, 8]; some quantitative differences exist, but there is substantial overlap between disease-gene groups, and exceptions are common [1, 2]. Additional complexities include the presence of two or more variants, as either compound or double heterozygosity (some families may have more than one causative mutation). The proportion of genotyped persons with more than one variant is higher in diseases with low penetrance — notably, ARVC. The presence of multiple variants complicates genetic testing in families [1, 2, 4, 8].
25.2 Hypertrophic Cardiomyopathy
25.2.1 Definition and Epidemiology
Hypertrophic cardiomyopathy (HCM) is defined as left ventricular (LV) hypertrophy in the absence of abnormal loading conditions sufficient to explain the degree of hypertrophy. HCM is a complex but relatively common form of genetic heart muscle disease that occurs in 1 out of 500 people, but often goes undiagnosed in the community. HCM is the most common cause of heart-related sudden death (SD) in people under 20 years of age, and it can also be responsible for exercise disability at almost any age. HCM occurs equally in both sexes and has been reported in many races (Fig. 25.3) [8–21].
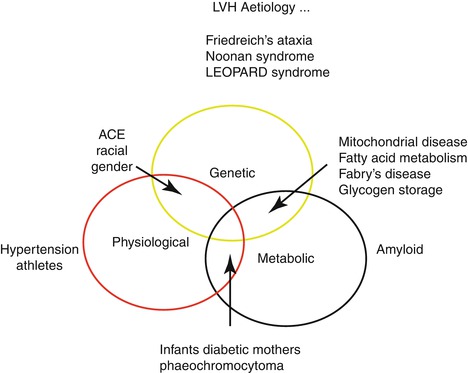
Fig. 25.3
LVH Aetiology
25.2.2 Genetics
HCM is a familial disease in approximately half of the cases. It is inherited mainly as an autosomal dominant trait caused by mutations in genes that encodes proteins of the cardiac sarcomere. The commonest are β-myosin heavy chain, cardiac troponin T, cardiac troponin I, α-tropomyosin, cardiac myosin binding protein C, the essential and regulatory myosin light chains, and cardiac actin. Other genes, such as those encoding α-myosin, titin, and proteins of the Z-disc, account for less than 1 % of cases (Table 25.1).
Table 25.1
Classification and causes of HCM
Familial | Nonfamilial |
|---|---|
Familial, unknown gene | Obesity Infants of diabetic mothers |
Sarcomeric protein disease | Athletic training |
β-Myosin heavy chain | Amyloid (AL/prealbumin) |
Cardiac myosin binding protein C | See Fig. 25.3 |
Cardiac troponin I | |
Troponin T | |
α-Tropomyosin | |
Essential myosin light chain | |
Regulatory myosin light chain | |
Cardiac actin | |
α-Myosin heavy chain | |
Titin | |
Troponin C | |
Muscle LIM protein | |
Glycogen storage disease | |
e.g., GSD II (Pompe disease); GSD III (Forbes disease); AMP kinase (WPW, HCM, conduction disease) | |
Danon disease | |
Lysosomal storage diseases | |
e.g., Anderson-Fabry disease, Hurler syndrome | |
Disorders of fatty acid metabolism | |
Carnitine deficiency | |
Phosphorylase B kinase deficiency | |
Mitochondrial cytopathies | |
e.g., MELAS, MERRF, LHON | |
Syndromic HCM | |
Noonan syndrome | |
LEOPARD syndrome | |
Friedreich ataxia | |
Beckwith-Wiedermann syndrome | |
Swyer syndrome (pure gonadal dysgenesis) | |
Costello syndrome | |
Other | |
Phospholamban promoter | |
Familial amyloid |
Non sarcomeric HCM refers to apparently similar disorders with different causes. Distinctions among such conditions can be clinically important, because disorders with similar cardiac morphology can have different inheritance patterns, natural histories, or responses to therapy.
25.2.3 Clinical Diagnosis
Most patients with HCM are asymptomatic. Some experience chest pain, breathlessness, fatigue, and palpitations, often with day-to-day variation. Syncope is also common and can be caused by many mechanisms including left ventricular outflow obstruction (LVOTO), hypotension secondary to abnormal vascular responses, and arrhythmias. Changes in LV loading conditions during exercise, heavy meals, and dehydration often precipitate symptoms. The cardiovascular examination in most patients is normal. On auscultation, LVOTO causes a harsh ejection systolic murmur at the left sternal edge, which increases in intensity during the strain phase of the Valsalva, or when standing from the squatting position. Most patients with LVOTO also have mitral regurgitation, caused by abnormal coaptation of the mitral valve leaflets during systole.
The resting 12-lead ECG is abnormal in 95 % of patients with HCM. Atrioventricular conduction abnormalities (including first-degree block) are rare, except in particular subtypes of HCM (e.g., PRKAG2 mutations, mitochondrial disease, Danon or Fabry disease) where they are more frequent (Fig. 25.3) [7, 9, 11].
25.2.3.1 Imaging
In patients with HCM, absolute left-ventricular wall thickness ranges widely from mild (13–15 mm) to massive (>50 mm) [4, 8, 15–19]. The diagnosis of HCM relies on the presence of a maximal LV wall thickness of more than 2 standard deviations from the normal (typically ≥13 mm in an adult). Hypertrophy is typically asymmetric, involving the interventricular septum in more than other segments, but any pattern of LV hypertrophy, including concentric, eccentric, distal, and apical, is consistent with the diagnosis of HCM. In resting conditions, 25 % of patients have obstruction of the LV outflow tract. As many as 70 % of symptomatic patients may have latent or provocable LV systolic function. A proportion of adults with HCM develop progressive myocardial thinning, global LV systolic impairment, and cavity dilatation. Characteristically, patients with HCM have diastolic LV impairment shown by reduced early diastolic (Ea) velocities in the mitral annulus and septum and reversal of the ratio of early/late diastolic velocities (Ea/Aa) (Fig. 25.4). Cardiac magnetic resonance imaging provides a detailed assessment of cardiac morphology as well as an accurate assessment of systolic function. It also permits tissue characterization, particularly detection of myocardial scarring by the assessment of delayed gadolinium enhancement. Studies suggest that the extent of gadolinium enhancement correlates with risk factors for sudden death and with progressive LV remodeling [4, 8, 16, 20].
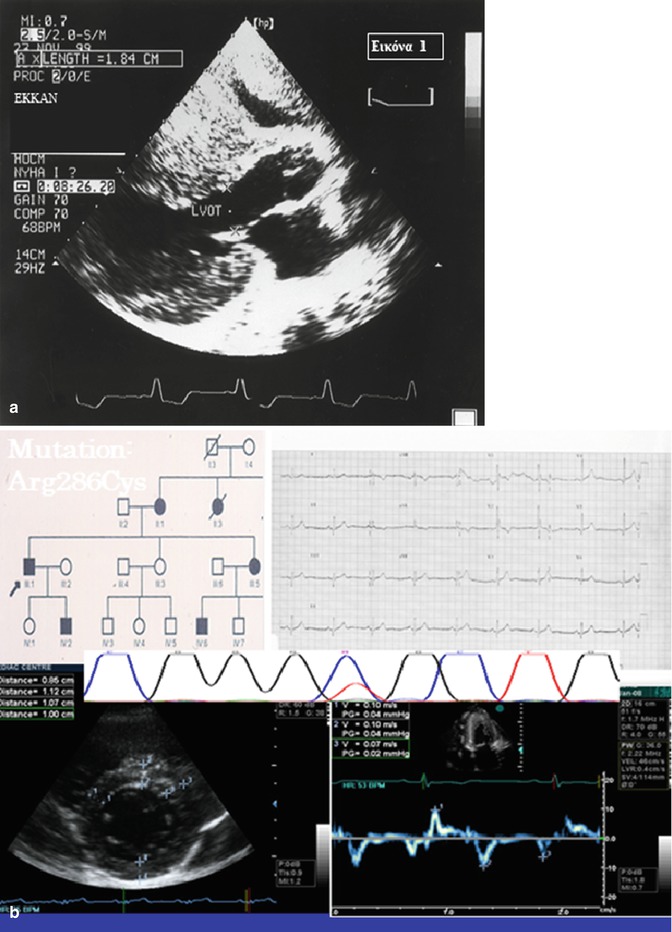
Fig. 25.4
Hypertrophic cardiomyopathy: (a) Typical form (b) Subclinical form (Image reproduced courtesy of EKKAN)
25.2.3.2 Further Investigation
Symptom limited exercise testing is safe in HCM [50, 51] and provides a quantitative assessment of a patient’s exercise tolerance, particularly when combined with metabolic functional testing. An abnormal blood pressure response to exercise is associated with an increased risk of SD in young adults. Ambulatory electrocardiographic monitoring is important in the assessment of symptoms and in the prediction of arrhythmic risk [21–48]. Nonsustained ventricular tachycardia occurs in 20 % of adults with HCM. Paroxysmal supraventricular arrhythmias occur in 30–50 % of patients; sustained atrial fibrillation (AF) is present in 5 % of patients at diagnosis, and develops in a further 10 % in the subsequent 5 years. The main indications for coronary angiography are exclusion of coronary artery disease and, much less commonly, assessment of cardiac output, filling pressures, and intraventricular pressure gradients in patients with severe symptoms. Endomyocardial biopsy is occasionally indicated when an infiltrative or metabolic disease, such as amyloidosis or Anderson-Fabry disease, is suspected [52–54].
25.2.4 Natural History
HCM can present at any age. Many patients follow a stable and benign course, with a low risk of adverse events, but a minority experience progressive symptoms caused by slow deterioration in diastolic and sometimes in LV systolic function. A proportion of individuals die suddenly, whereas others may die from progressive heart failure, thromboembolism, and, rarely, infective endocarditis [55].
25.2.4.1 Sudden Cardiac Death
Overall, HCM is a relatively benign disease but at the same time is one of the commonest causes of SCD in the young. Most contemporary studies report an annual incidence of SCD in HCM populations of 0.5–1 % per year, rising to 2 % or higher in certain groups. The mechanism of SCD is rarely documented but factors that contribute to a propensity to ventricular arrhythmias include dispersion of repolarization, which increases susceptibility to triggered arrhythmias; myocyte disarray and areas of conduction block that predispose to reentry arrhythmias. Other morphologic and physiologic factors influence the vulnerability of the underlying substrate, such as myocardial ischemia, LVOTO, and diastolic dysfunction [4, 8, 38–61].
25.2.4.2 End Stage HCM, Stroke and Infective Endocarditis
End-stage HCM develops at all ages but, in most patients, the time from onset of heart failure (HF) symptoms to diagnosis of severe systolic impairment is about 10–15 years. The development of severe systolic HF is associated with a poor prognosis, with rapid progression to death or transplantation and an overall mortality of up to 11 % per year. The prevalence of severe systolic impairment in HCM using conventional echocardiographic criteria ranges from 2 % to nearly 10 %, with an annual incidence of less than 1 %. The annual incidence of stroke varies from 0.56 to 0.8 %/y, rising to 1.9 % in patients more than 60 years old. The major cause is atrial fibrillation [AF], which affects about a quarter of patients with HCM, with an incidence of 2 % per annum. Risk factors for AF include age and left atrial dilation (a consequence of diastolic dysfunction, LVOTO, and mitral regurgitation). Patients with obstructive HCM have an increased risk of developing infective endocarditis, usually on the anterior mitral valve leaflet. The incidence of infective endocarditis is 1.4 per 1,000 person-year and 3.8 per 1,000 person-year in patients with obstruction [55, 56].
25.2.5 Clinical Μanagement of HCM
The treatment of most patients with HCM focuses on the management of symptoms, the prevention of disease-related complications and the counseling of family members. Exceptions include lysosomal storage diseases, such as Pompe, Anderson-Fabry disease or amyloidosis, for which specific therapies are available (Figs. 25.3 and 25.5).
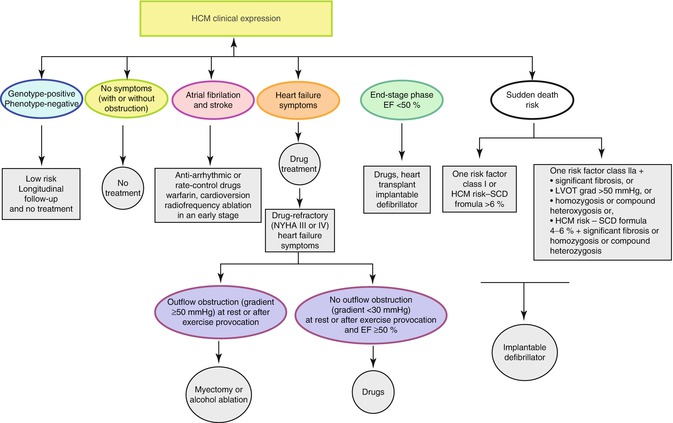
Fig. 25.5
Natural history and management of Hypertrophic Cardiomyopathy (Image reproduced courtesy of EKKAN). LVOT Left ventricular outflow track, EF Ejectiar fraction, NYHA New york heart association
25.2.5.1 Symptom Management
In patients with symptoms caused by LVOTO, the aim of treatment is to reduce the outflow tract gradient. Options include negative inotropic drugs (β-blockers, disopyramide, and verapamil), atrioventricular sequential pacing, percutaneous alcohol ablation of the interventricular septum, and surgery. Surgery or septal ablation is considered in patients with significant outflow obstruction (gradient >50 mmHg) and symptoms refractory to medical therapy [62, 63]. Therapeutic options in patients without LV outflow gradients are limited predominantly to pharmacologic therapy. Anticoagulation should be considered in all patients with sustained or paroxysmal AF [58, 59].
25.2.5.2 Risk Stratification and Prevention of Sudden Cardiac Death
SCD occurs throughout life, with a maximal incidence in adolescence and young adulthood, often without warning signs or symptoms. Although there is an excess of deaths during or after strenuous exertion, most occur during mild exertion or sedentary activities. The mechanism underlying most cases of SCD is believed to be ventricular tachyarrhythmia, but conduction disease and thrombo-embolism may account for some cases.
In general, the presence of one or more conventional clinical risk factors should prompt consideration of an implantable cardioverter defibrillator (ICD). Patients with two or more risk factors have a 4–5 % annual risk of SCD. More problematic are patients who have only a single risk factor (up to 25 % of patients). When assessing the need for ICDs in this group, age, symptoms, and the presence of so-called minor risk factors should be taken into account and balanced against the risk of complications of ICD therapy, particularly in young people. Patients with HCM should refrain from intense exercise. This restriction has significant implications for all patients, underlining the importance of accurate diagnosis [42–48].
25.2.5.3 Clinical Risk Stratification in HCM
The Following Data are Being Used
1.
Conventional primary prevention risk markers
Family history of sudden death due to hypertrophic cardiomyopathy
Unexplained recent syncope
Multiple repetitive non-sustained ventricular tachycardia (on ambulatory ECG)
Hypotensive or attenuated blood pressure response to exercise
Massive left-ventricular hypertrophy (thickness, ≥30 mm)
2.
Potential high-risk subsets for primary prevention
End-stage phase (ejection fraction <50 %)
Left-ventricular apical aneurysm and scarring
3.
Potential arbitrators for primary prevention1
Substantial left-ventricular outflow gradient at rest
Multiple sarcomere mutations
Extensive and diffuse late gadolinium enhancement
Modifiable
Intense competitive sports
Coronary artery disease
Recently a more sophisticated model (HCM Risk-SCD) was published for predicting SCD in HCM. This is the first validated SCD risk prediction model for patients with HCM. The model provides accurate individualized estimates for the probability of SCD using readily collected clinical parameters such as LVH, LVOT gradient at rest, NSVT in Holter, familial SCD, LA size and age [47].
25.2.5.4 Clinical Value of Genetic Testing
A genetic diagnosis is obtainable in 70 % of consecutive patients with familial HCM, though the yield is lower (30 %) when sporadic disease is considered [4, 8]. Those with special characteristics such as conduction disorders, pre-excitation, or systemic disease may represent phenocopies of sarcomere-associated HCM and focused genetic testing may be helpful with diagnosis and treatment (e.g. Danon’s disease, Fabry disease). The greatest benefit of genetic testing in HCM derives from cascade screening with the ability to identify which individuals in a family are, or are not, at risk of disease development [1, 2, 4, 5, 8, 32, 60, 63, 64].
25.2.6 Evaluation of Families and Genetic Counselling
Current guidelines recommend screening with a 12-lead ECG and echocardiogram for family members of HCM patients [4, 5, 8, 63].
At age <12 years
Screening optional unless:
Either a malignant family history of premature death from hypertrophic cardiomyopathy is known or other adverse complications are present
Child is a competitive athlete in an intensive training programme
Onset of symptoms
Other clinical suspicion of early left-ventricular hypertrophy has been noted
At age 12–21 years
Screening should be performing every 12–18 months
At older than 21 years
Imaging should be performed either at onset of symptoms or possibly at 5-year intervals (at least through mid-life); more frequent intervals are appropriate in families with a malignant clinical course or history of late-onset hypertrophic cardiomyopathy
Thus at the present time hypertrophic cardiomyopathy has been transformed from a rare and largely untreatable disorder to a common genetic disease with effective management strategies.
25.3 Arrhythmogenic (ARVC/ALVC) Cardiomyopathy
25.3.1 Definition-Epidemiology
Arrhythmogenic right ventricular cardiomyopathy (ARVC) is a genetically determined heart muscle disease that predominantly affects the right ventricle (RV). The disease is characterized by fibrofatty replacement of the right and/or the left ventricular myocardium, segmental dysfunction of the RV or/and LV and arrhythmias. ARVC is mainly a desmosomal disease resulting from defective cell adhesion proteins. The estimated prevalence of ARVC in the general population ranges from 1 in 2,000 to 1 in 5,000. A familial background has been demonstrated in about 50 % of ARVC cases (Fig. 25.6). The disease becomes clinically overt most often in the third or fourth decade of life. Clinical diagnosis of ARVC is often difficult because of the nonspecific nature of the disease and the broad spectrum of phenotypic manifestation, ranging from severe to concealed forms. In 2010, the International Task Force proposed revised criteria for the clinical diagnosis of ARVC (Table 25.2) [65].
Table 25.2
Clinical diagnostic criteria for right versus left arrhythmogenic cardiomyopathy (ACM)
Diagnosis ACM | 12-Lead ECG | Signal-averaged ECG | Arrhythmia | Ventricular volumes | RV/LV volume ratio | Other imagine abnormalities | |
|---|---|---|---|---|---|---|---|
Classic right dominant ARVC | Normal | Late potentials | Both supraventricular tachycardia and atrial fibrillation/flutter are observed but are not contributory to diagnosis | Frequent PVCs of LBBB configuration | Normal | ≥1.2, increases with disease progression | Localized dilation, WMA, and/or aneyrysms in RV, preferentially affecting triangle of dyplasia and mid-free wall |
Poor R-wave progressiona | Sustained/non-sustained VT of LBBB configurationb | Mild, moderate, or severe RV dilation ± dysfunction | Increased/abnormal trabeculation | ||||
IVCD in V1-3 | Fat/late enhancement in RV myocardium | ||||||
Incomplete RBBB | |||||||
RBBB | |||||||
Epsilon waves in V1-3 | |||||||
Inverted/flat T-waves in V1-3, extending to V4-6 with LV involvement | |||||||
ST elevation in V1-3 | |||||||
Left dominant ALVC | Normal | – | Frequent PVCs of RBBB configuration | Normal | <1, diminishes with disease progression | Localized dilation, WMA, and/or aneyrysms in LV | |
Leftward QRS axis (−30°< QRS axis <0) or left-axis deviation (QRS axis <−30°) | Sustained/non-sustained VT of RBBB configurationc | Mild, moderate, or severe LV dilation ± dysfunction | Non-compacted appearance | ||||
Early transition | Late enhancement in LV myocardium in subepicardial/midwall distribution | ||||||
LBBB | |||||||
Epsilon waves in inferior (II, III, aVF) and/or lateral leads (V5-V6 ± V4, I, aVL) | |||||||
Inverted/flat T-waves in (infero) lateral leads, extending to V1-3 with RV involvement | |||||||
25.3.2 Genetics
The disease is familial, and typically autosomal dominant, in about half the cases. Mutations in five genes that encode desmosomal proteins (desmoplakin, plakoglobin, plakophilin 2, desmoglein 2, and desmocollin 2) have been found in ARVC. The same holds true for two related autosomal recessive disorders: (1) Naxos disease (ARVC accompanied by woolly hair and palmoplantar keratoderma) and, (2) The Carvajal syndrome (which has a similar dermatologic phenotype but in which left ventricular involvement is predominant). Mutant desmosomes may therefore compromise cell-to-cell adhesion at intercalated disks, lessening the ability of myocytes to withstand mechanical forces during the cardiac cycle. Damage to the cell surfaces, causing cell detachment and cell death, probably ensues [65, 66].
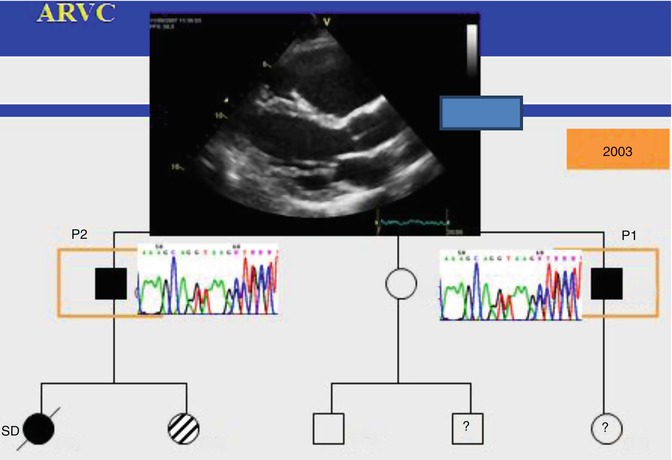
Fig. 25.6
ARVC is a familial cardiomyopathy (Image reproduced courtesy of EKKAN)
The majority of causative mutations are insertions or deletions or nonsense mutations that result in premature truncation of the encoded proteins. Two other, nondesmosomal genes have been implicated in ARVC: one for the transforming growth factor β3 (TGF-β3) and the other for the transmembrane protein 43 (TMEM43). Experimental data indicating that mutant desmosomes also cause remodeling of gap junctions 63 explain how electrocardiographic changes and ventricular arrhythmias can develop before the loss of myocytes and dysfunction of the RV become apparent (the concealed phase of the disease) [65–71].
25.3.2.1 Morphologic Types of ACM
The spectrum of RV alterations ranges from global RV dilatation/dysfunction to regional wall motion abnormalities or “bulges” typically localized in the triangle of dysplasia, (i.e., subtricuspid, apical, and infundibular regions). The LV and the septum are usually involved to a lesser extent; whereas, biventricular or left dominant variants of disease have been reported [71–76].
The extent of LV involvement and variations in the perceptible timing has allowed recognition of three distinct patterns of disease expression:
1.
Classic (right dominant). The early stages of the disease are characterized by isolated RV involvement, evolving from localized to diffuse. LV involvement may arise after the onset of global RV dysfunction.
2.
Left dominant, characterized by early prominent LV involvement; the LV is consistently more severely affected than the right.
3.
Biventricular, characterized by parallel bilateral involvement with no apparent proclivity to either ventricle.
Left-dominant arrhythmogenic cardiomyopathy (LDAC) is characterized pathologically by fibroadipose replacement of the LV. In classic right dominant disease, LV involvement is frequently confined to the inferolateral and inferior walls, sparing the septum. LDAC, in contrast, affects the septum in more than 50 % of cases. Cardiovascular magnetic resonance commonly shows late gadolinium enhancement in a subepicardial or midmyocardial distribution, consistent with that observed in pathology [74].
25.3.3 Clinical Diagnosis
25.3.3.1 Original and Modified Task Force Criteria
The 1994 Task Force Criteria were initially designed to guarantee an adequate specificity for ARVC among index cases with overt clinical manifestations. However, the 1994 Criteria lack sensitivity for identification of early/minor phenotypes, particularly in the setting of familial ARVC. Accordingly, diagnostics criteria have been recently revised with the aim of improving diagnostic sensitivity [65]. Comparison of dignostic criteria of ARVC versus ALVC [LDAC] is presented in Table 25.2. The approach of classifying structural, histopathologic, electrocardiographic (ECG), arrhythmic, and genetic features of the disease as major and minor criteria has been maintained. As far as ECG and arrhythmic features are concerned, in the revised Task Force Criteria T-wave inversion in V1 to V3 as well as VT with left bundle branch block (LBBB) morphology with superior/indeterminate QRS axis, either sustained or non-sustained, have become major criteria. The following findings have been included among minor criteria: (1) T-wave inversion in V1 and V2 in the absence of right bundle block (RBBB) and from V1 to V4 in the presence of complete RBBB; (2) Positivity of any one of the three signal-averaged ECG (SAECG) parameters for late potentials; and (3) premature ventricular beats more frequent than 500 per 24 h on the Holter monitoring. Moreover, revised guidelines provide quantitative cutoff values in imaging and histopathological criteria. Finally, the identification of a pathogenetic gene mutation in a first-degree relative has become a major criterion for ARVC diagnosis [65, 71, 74, 76].
Table 25.3
Definition of the clinical status of members of families with familial dilated cardiomyopathy compiled from [83]
Major criteria | Minor criteria | |
1. The presence of two or more affected individuals in a single family; or | 1. Unexplained supraventricular (atrial fibrillation or sustained arrhythmias) or ventricular arrhythmias, frequent (>1,000/24 h) or repetitive (three or more beats with >120 bpm) before the age of 50 < div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

| |