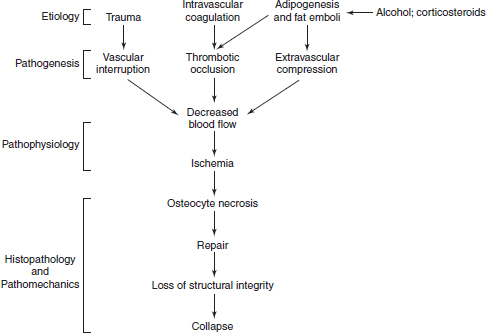
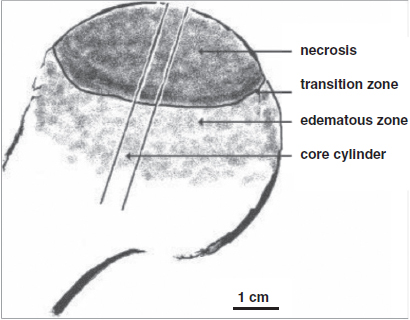
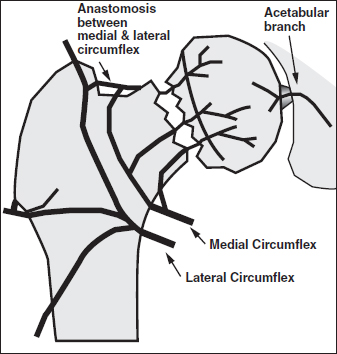
CHAPTER 7 The clinical features, etiology, and natural history of osteonecrosis (ON) have been well described.1, 19, 60, 66, 98 This chapter focuses, and has a distinct emphasis, on circulatory pathologies and puts them in the context of the histopathology and pathomechanics of bone necrosis, subchondral resorption, subsequent subchondral fracture, collapse, and joint incongruity. ON has typically been classified by etiology and pathogenesis, and since most relevant studies of pathophysiology have been in late-stage human ON, combinations of pathologic observations and uncertainty as to their temporal sequences cloud the ability to finely parse out inciting primary and secondary events.47, 63, 76 Various putative etiological conditions converge on a more or less stereotypical pathophysiology that reflects circulatory compromise. A unifying concept of the pathogenesis of ON emphasizes the role of circulatory occlusion leading to ischemia and osteocyte necrosis (Fig. 7.1). Many etiologies may contribute to the pathogenic mechanisms of vascular interruption, thrombotic occlusion, or extravascular compression, any of which may decrease perfusion and result in ischemia and necrosis.1 It has been convenient to subdivide ON as traumatic and non-traumatic and to further subdivide non-traumatic ON as resulting from primarily intravascular pathology (e.g., hypercoagulation and thrombosis) or primarily extravascular pathology (e.g., adipocyte hypertrophy, Gaucher disease). Fig. 7.1. A concept of pathogenesis of ON that unifies several etiologic and pathogenic hypotheses. Source: Aaron RK, Gray R. Osteonecrosis: Etiology, natural history, patho-physiology and diagnosis. In: Callaghan JJ, Rosenberg AG, Rubash HE, eds., The Adult Hip. Philadelphia, PA: Lippincott Williams, 463–476. Reprinted with permission from Wolters Kluwer Health. The pathologic features of ON focusing on vascularity are presented in this book in Chapter 8 and elsewhere.1 In summary, the presence of necrotic bone induces a repair process in which bone resorption exceeds formation, leading to a loss of structural integrity in the subchondral trabeculae and, eventually, to subchondral collapse.25–27 It is not the necrosis per se but rather the repair process dominated by bone resorption that leads to the loss of subchondral bone.1 Finite element modeling has demonstrated that the resorption of subchondral trabeculae is the major contributor to the loss of structural integrity and eventual collapse and incongruity of the articular surface.9 Fig. 7.2. Schematic view of ON femoral head showing necrotic, transitional, and edematous zones. Source: Radke, et al. (2006). Expression of the angiomatrix and angiogenic proteins CYR61, CTGF, and VEGF in osteonecrosis of the femoral head. J Orthop Res, 24(5): 945–952. Reprinted with permission. The histopathology of ON has been well described.8, 10, 16, 19, 43, 88 The necrotic area, usually encompassing the weight-bearing area of the femoral head, is characterized by absence of bone cells and necrotic marrow. It is described as containing granulation tissue reminiscent of caseating necrosis and containing saponified fat. Depending on the progression of the disease, it is often surrounded by a zone of repair with evidence of creeping substitution (new bone overlaying on dead bone) — sometimes called a rim of sclerosis. Beneath therepair zone is a hyperemic zone containing numerous blood vessels (Fig. 7.2). The histopathology of ON has been categorized by Ficat and Arlet as fourtypes:19 type 1 (prenecrotic): plasmostasis, reticular proliferation, hemorrhage, accumulation of foam cells; type 2 (preradiologic): necrosis of fatty marrow; type 3: medullary necrosis with evidence of 50–100% of the trabecular lacunae empty; type 4: necrosis and fibrosis with foci of necrosis surrounded by new bone.5, 36–38 Saito et al. described the histology of core biopsy specimens identifying four zones ranging from complete necrosis with ischemia (zone A) to transitional zones with necrosis containing evidence of old (zone B) or new hemorrhage (zone C) to intact bone (zone D) with living fatty or hematopoietic tissue.82 A definitive diagnosis can be made when “creeping substitution” — new bone overlying necrotic bone — is detected.8, 41 The blood vessels that supply the femoral head include the medial and lateral circumflex, a vessel of the ligamentum teres, and the inguinal gluteal arteries. The medial circumflex and lateral circumflex arteries form a vascular ring around the femoral neck.44, 55, 113 Two sets of retinacular vessels branch off from this ring including (1) metaphyseal branches which penetrate into the femoral neck and (2) epiphyseal branches which penetrate into the peripheral non-articular portion of the head to supply the epiphysis. Approximately 26 lateral epiphyseal arteries penetrate the femoral head posteriorly and superiorly supplying the lateral weight-bearing portion of the femoral head.44 While it has also been shown that the inferior gluteal artery contributes significantly to the blood supply to the femoral head of developing fetuses, its role is less clear in adults.113 The vessels of the ligamentum teres also carry a small supply to the femoral head, but its contribution to its blood supply is considered to be minimal.44, 113 Veins are more difficult to visualize because venous valves may block retrograde extraosseous injections and veins are thinner walled and more collapsible than their arterial counterparts.55 Epiphyseal and metaphyseal veins are more numerous than their corresponding arteries.44 Veins within the medullary space, termed marrow sinusoids, drain through collecting ducts into the central venous sinus; a branch of the central venous sinus, the nutrient vein, passes through the nutrient foramen along with the nutrient artery.44, 55 Several different pathologic mechanisms that may result in ischemia and subsequent lesions of ON include vascular disruption, intraosseous extravascular compression, extraosseous vascular constriction, and intravascular occlusion.110 The anatomy and location of the vessels to and from the femoral head make them especially vulnerable to trauma (Fig. 7.3). Trauma that compromises the femoral neck may compromise the vascular ring around the neck and the circulation to and from the femoral head. There is a notable incidence of ON with displaced and undisplaced femoral neck fractures, ranging from 14.1 to 100%.13 ON has also been reported to occur in association with hip dislocations (3% anterior; 13% posterior).13 Fig. 7.3. Disruption of ascending vasculature and devascularization of the femoral head as a consequence of femoral neck fracture. Source: Johnson EO, Soultanis K, Soucacos PN (2004). Vascular anatomy and microcirculation of skeletal zones vulnerable to osteonecrosis: Vascularization of the femoral head. Orthop Clin North Am, 35(3), 285–291, viii. Reprinted with permission from Elsevier. Using superselective angiography, Atsumi and Kuroki identified five types of distributions of the superior retinacular arteries.6 In normal cases, the superior retinacular arteries were characterized as arch type and extended to the center of the femoral head (type 1). The other types included(1) failure to visualize vessels due to extraosseous pathology (type 2) and(2) various degrees of small arterial penetration (types 3–5). The majority of the hips with early-stage necrosis demonstrated type 3 findings (the presence of small arteries penetrating where the superior retinacular arteries were normally located). The authors suggested that this represents revascularization. These findings were also noted in the contralateral “normal” hips and in normal hips with steroid administration. Further study is needed to determine whether these abnormalities existed before or after ON had been initiated. Serre and Simonfound normal femoral arteriograms with abnormal intraosseous phlebography in patients with ON of the femoral head.86 Hungerford observed absent or incomplete filling of the main extraosseous veins, diaphyseal reflux, and stasis of contrast media on 5-min and subsequent films.37 Prior to MRIs, core biopsies and intraosseous pressure measurements were often performed in order to confirm a diagnosis of ON. Examination of the core specimens indicated that there were increased numbers of adipocytes and adipocyte hypertrophy, frequently associated with high dose corticosteroid administration. Another finding was that patients with ON frequently exhibited elevated intraosseous pressures and abnormal stress tests.19, 37, 45 A stress test involved the administration of a 5-ml bolus of saline into the head, creating a temporary increase in pressure. If normal, the pressure decreased rapidly following injection; if abnormal, the increase returned more slowly to pre-injection levels. Based upon these findings, it was suggested that ON developed as a consequence of compression of the thin-walled veins by encroaching adipocytes resulting in venous stasis.39 This hypothesis was supported by observations of abnormal venograms. The femoral head is a rigid compartment with marrow elements located within the spaces of trabecular bone. If there is an increase in intraosseous extravascular pressure, perfusion could be theoretically impaired. Intraosseous extravascular occlusion and intraosseous hypertension can conceivably occur if there is an increase in the adipocyte number and/or hypertrophy, or with increased numbers of Gaucher cells. Some experimental observations have reported the presence of intraosseous hypertension70, 101 but others have found that the measurement of intraosseous pressure lacks sufficient diagnostic precision, being neither sensitive nor specific.1 Various observations have led to the conclusion that intraosseous hypertension, while often seen in ON, is a non-specific and secondary, perhaps contributory, but not causal, factor in ON.49, 50 Although the patho-physiology is not definitively known, intraosseous, space-occupying factors are clinically associated with ON. Lipid deposition and lipocyte hypertrophy have been observed in specimens of ON bone, often associated with corticosteroid or alcohol intake.54, 91, 102 It has been hypothesized that accumulating lipid and hypertrophic cells create extravascular pressure and decrease either arterial inflow or venous outflow, or both. Alcohol- or corticosteroid-associated adipogenesis may shift precursor cells from an osteocytic to an adipogenic lineage, thereby reducing the number of osteoprogenitor cells.105 Gaucher disease is associated with a high incidence of ON.17 Gaucher cells are specialized cells that accumulate large amounts of glucocerebrosides, a type of glycosphingolipid. In Gaucher disease, there is substantial extravascular medullary infiltration by Gaucher cells resulting in an increase in intracortical pressure. It has been suggested that this increased pressure within the rigid compartment of the femoral head results in compression of the vascular sinusoids leading to bone ischemia.85 This is explored further in Chapter 8. Impaired venous outflow (venous stasis) has been observed to increase intraosseous pressure, reduce perfusion, and result in hypoxia, and has been proposed as a cause of ON.38, 104 Extraosseous venous obstruction has been implicated as a cause of venous stasis.20 Hypoxia and reduced intraosseous fluid flow have been measured in the subchondral bone of hips with non-traumatic ON, suggesting a causal role in osteocyte necrosis.20 A number of laboratory studies have investigated the response of the endothelium and the effect on blood flow of ischemia following the clamping off of extraosseous blood vessels.14, 97, 106 Ischemic skeletal muscle results in diminution of arterial diameters followed by active constriction, forcing blood elements distally and resulting in a reduced or even completely occluded lumens.14 This study noted swelling of endothelia and leukocytes, erythrocyte rouleaux formation, and distortion of leukocytes and erythrocytes during ischemia. During reperfusion, alteration of flow patterns, vortex formation, regional stasis, focal hemorrhage, edema, vasospasm, platelet aggregation, erythrocyte sludging, adhesion and migration of leukocytes, and swelling of endothelia and leukocytes were observed. Winetet al. used intravital microscopy to study blood flow to bone.36, 106 Using a model where they were able to clamp the right common iliac artery in rabbits, they were able to create ischemia to bone. Upon release of the clamp, they observed secondary ischemia/no-reflow and other evidence of reperfusion injury was observed.36 This was associated with abnormally high leakage of FITC-D70 from the few vessels perfused during secondary ischemia, indicating that damage to the blood vessels had occurred. Further study is needed to investigate the possibility of reperfusion injury associated with ON; this has clinical ramifications regarding both an understanding of the pathogenesis and treatment of ON. Intravascular occlusion may result from thrombosis, fat or gas embolism, or sickle cell aggregation. Fat emboli have been observed in cases of ON. Jones observed fat emboli in patients with dysbaric ON and following an allergic reaction to an insect bite, trauma, and alcohol.46 He reported that “there were arterioles, capillaries, and sinusoids occluded by deformed fat emboli and fibrin thrombi”.46 There is a strong association between sickle cell disease and ON.69 The sickling of blood cells may lead to repeated ischemic events related to intravascular obstruction.67, 85 In adolescents with sickle cell disease, Hernigou and Bernaudin reported that the blood flow in the sinusoidal system was sluggish and the biochemical environment in this area facilitated the sickling process; blood containing sickled cells had high viscosity and produced a relative obstruction to blood flow in the sinusoidal system of the epiphysis.33 This differs from other descriptions of ON in that the trabecular bone becomes necrotic while the marrow elements remain viable.85, 91 Furthermore, there is no blockage to revascularization and there is evidence of many cement lines indicating multiple attempts to heal the bone.85 This is explored further in Chapter 8. In spite of our knowledge about the relationship between dysbarism (Caisson disease) and ON, ON continues to occur in as many as 34.5% of individuals exposed to compressed air in 2014.62 With dysbarism, there is evidence of lipid dysregulation, coagulation abnormalities, and increased intracortical pressure.76 Hutter et al. proposes that the ON is a consequence of intravascular obliteration by nitrogen bubbles or microthrombi or ofextraluminal obliteration by bone marrow gas or edema.40 Another concept is that bubbles within the fatty marrow cause an increase in intramedullary pressure, occluding the intraosseous and extraosseous vessels, thereby leading to stasis of blood and subsequent ischemia of osteocytes.87 J.P. Jones observed multiple gas bubbles within the large vessels and the fatty marrow of femoral and humeral heads associated with lipid and platelet aggregates on the surface of marrow bubbles. He also noted fibrin-platelet thrombi within dilated venous sinusoids adjacent to bubbles, and in veins, capillaries, and arterioles associated with intraosseous fat embolism and fibrin thrombi in the subchondral bone.48 He proposed that injured marrow adipocytes can release liquid fat, thromboplastin, and other vasoactive substances that serve as procoagulants and trigger disseminated intravascular coagulation and ON. Unlike secondary and idiopathic ON of the femoral head, the regions affected by dysbarism may include the head, neck, and proximal shaft of the femur and humerus.87 Intravascular occlusion resulting from thrombi within the microvascular circulation has been implicated in the development of ON of the femoral head. Thrombi may form in response to endothelial damage or dysfunction.56 The consequences of thrombi range from a diminution of the blood flow through larger vessels to complete occlusion of smaller blood vessels. Evidence of endothelial damage and dysfunction in the bone of patients with ON include sinusoidal distension, thickening of the arteriolar walls, arteriolar thrombosis and fibrin thrombi in the subchondral Haversian capillaries, arterioles, and marrow sinusoids.19, 47 Thrombi have been detected in both intraosseous arterioles and venules.84, 92 Evidence of old and new intramedullary hemorrhage has also been observed.47, 82 Thrombi have been frequently observed when the inciting event for the ON is known (e.g., dysbaric ON). In studies that are investigating a possible genetic predisposition in patients with ON, there has been a focus on the genes associated with hypofibrinolysis and thrombophilia.28, 31, 51, 111 Both disorders result in increased thrombi and blood flow impairment. Glueck et al. has observed that 4G/4G polymorphism for the gene for PAI-1 was associated with ON and that the gene product, PAI-Fx, was high in 31% of the ON patients as compared to the control subjects (3%).29 With respect to thrombophilia, there is evidence of mutations of the genes for methylenetetrahydrofolate reductase (MTHFR), Factor V Leiden, and prothrombin. For example, Zalavras et al. reported that there was a significantly higher percentage of ON patients (18%) with the G1691A mutation of factor V Leiden as compared to controls (4.6%).111 Elevated coagulation factors can also be found in ON patients without evidence of genetic mutations. This acquired hypercoagulability may be related to other co-morbidities or to pharmaceutical treatment (for example, corticosteroids). Jones et al. found that 82.2% of ON patients had at least one abnormal coagulation factor compared to 30% of healthy controls.52 Furthermore, almost 50% of the patients had two or more abnormalities as compared to 2.5% of the healthy controls. Regarding idiopathic ON (no identifiable risk factors), Jones et al. found at least one coagulation factor abnormality in the sera of all five patients in their study; high aCL IgG and PAI-1 were evident in four patients. Zalavras et al. reported low protein C, low protein S, high lipoprotein(a), and high von Willebrand factor levels present in a significantly higher proportion of patients with idiopathic ON (59%) and secondary ON (63%) compared with control subjects (8%).109 In the same patient population, Cenni found higher plasminogen and D-dimer levels and lower protein C levels in patients with idiopathic ON than in healthy controls.12 Several investigators have studied the vasculature contained within and around ON lesions. In an examination of core biopsy specimens, Arlet et al. reported fewer thick- and thin-walled blood vessels as compared to samples from patients diagnosed with osteoarthritis or algodystrophy.4 They observed fibrosis and thickening of the media with narrowing of the lumen. In a few of the cases, they found evidence of intravascular thrombosis. They also detected parietal ruptures and hemorrhages within the sinusoids. One limitation of this study is that there is no description of where within the affected tissue the vessels were located. Saito et al. reported that the number of blood vessels in the bone marrow was reduced in the necrotic core biopsies.83
CIRCULATORY PATHOLOGY
IN OSTEONECROSIS
7.1 Introduction
7.2 Histopathology and Pathomechanics
7.3 Vasculature and Vascular Pathology
7.3.1 Vascular disruption
7.3.2 Intraosseous extravascular compression
7.3.2.1 Lipid accumulation and lipocyte hypertrophy
7.3.2.2 Gaucher disease
7.3.3 Extraosseous vascular constriction
7.3.4 Intravascular occlusion
7.3.4.1 Sickle cell anemia
7.3.4.2 Dysbarism
7.3.4.3 Hypercoagulation and thrombosis
7.4 Circulatory Pathophysiology and Vascular Pathology
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


