Chapter 41 Chronic Obstructive Pulmonary Disease
Epidemiology, Pathophysiology, and Clinical Evaluation
Definitions and Diagnostic Considerations
The diagnosis of COPD should be considered in any person with the following:
• A history of chronic progressive symptoms: cough, wheeze, and/or breathlessness, with little variation in these symptoms
• A history of exposure to risk factors: cigarette smoke, occupational and environmental dust, and gaseous exposure
Pathology
Although the clinical and physiologic presentation of chronic asthma may be indistinguishable from COPD, the pathologic changes are distinct from those in most cases of COPD, largely because of cigarette smoking. The histologic features of COPD in the 15% to 20% of COPD patients who are nonsmokers have not yet been studied in detail. Although complex, the pathology of COPD can be simplified by considering separate disease sites in which pathologic changes occur in smokers to produce a clinical pattern of largely fixed airflow limitation (Box 41-1). The clinicopathologic picture is complicated because chronic bronchitis, bronchiolitis, and emphysema may exist in an individual patient, resulting in the clinical and pathophysiologic heterogeneity seen in patients with COPD.
Box 41-1 Chronic Obstructive Pulmonary Disease (COPD)
Pathologic Changes
Proximal Airways (cartilaginous airways >2 mm in diameter)
Macrophages and CD8 T lymphocytes
Few neutrophils and eosinophils (neutrophils increase with progressive disease)
Submucosal bronchial gland enlargement and goblet cell metaplasia (results in excessive mucus production or chronic bronchitis)
Cellular infiltrates (neutrophils and lymphocytes) of bronchial glands
Airway epithelial squamous metaplasia, ciliary dysfunction, increased smooth muscle and connective tissue
Peripheral Airways (noncartilaginous airways <2 mm in diameter)
Macrophages and T lymphocytes (CD8+ > CD4+)
Few neutrophils or eosinophils
Pathologic extension of goblet cells and squamous metaplasia into peripheral airways
Luminal and inflammatory exudates
B lymphocytes, lymphoid follicles, and fibroblasts
Peribronchial fibrosis and airway narrowing with progressive disease
Lung Parenchyma (respiratory bronchioles and alveoli)
Macrophages and CD8+ T lymphocytes
Alveolar wall destruction caused by loss of epithelial and endothelial cells
Development of emphysema (abnormal enlargement of air spaces distal to terminal bronchioles)
Microscopic emphysematous changes
Macroscopic emphysematous changes
Chronic Bronchitis
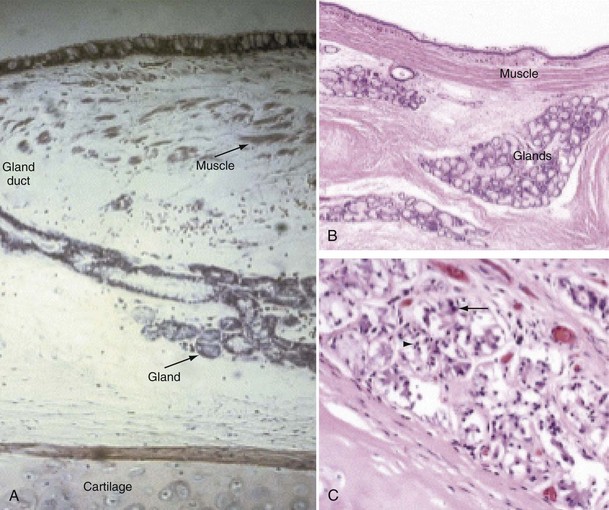
Mucus is produced by mucous glands present in the larger airways and by goblet cells in the airway epithelium. Chronic bronchitis is characterized by hypertrophy of the mucous glands (Figure 41-1). Goblet cells that occur predominantly in the surface epithelium of the larger airways increase in number and change in distribution, extending more peripherally. Bronchial biopsy studies confirm findings in resected lungs and show bronchial wall inflammation in chronic bronchitis. Activated T lymphocytes are prominent in the proximal airway walls, with a predominance of the CD8 suppressor T lymphocyte subset, rather than the CD4 subset, as seen in asthma. Macrophages are also prominent. Sputum volume correlates with the degree of inflammation in the airway wall. Neutrophils are present, particularly in the bronchial mucus-secreting glands (Figure 41-1), and become more prominent as the disease progresses. In stable chronic bronchitis, the high percentage of intraluminal neutrophils is associated with the presence of neutrophil chemotactic factors, including interleukin-8 (IL-8) and leukotriene B4 (LTB4). Elastase released from these cells is a potent stimulant for the secretion of mucus. Macrophages and CD8+ T cells also accumulate in the mucous glands.
Small Airways Disease and Bronchiolitis
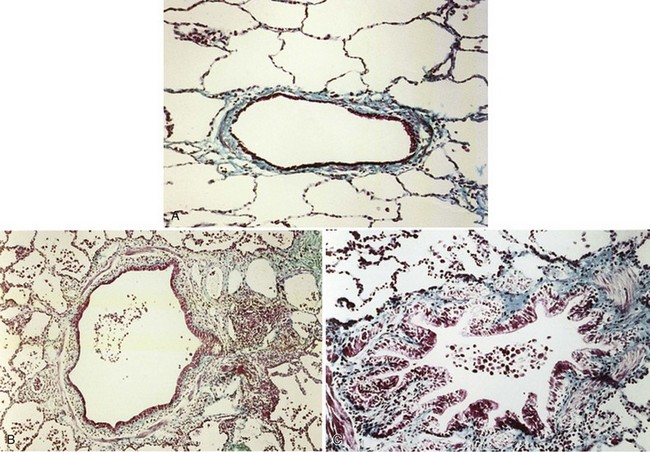
The smaller bronchioles (<2 mm in internal diameter) normally contribute relatively little to the total airway resistance, because there are so many airways of this size in parallel. Considerable narrowing of these airways can occur before pulmonary function becomes impaired and symptoms develop. Small airways inflammation is one of the earliest changes in asymptomatic cigarette smokers. The inflammatory cell profiles in the small airways are similar to those in larger airways, including the predominance of CD8+ lymphocytes, increase in CD8/CD4 ratio, and increased macrophages. Mucosal ulceration, goblet cell hyperplasia, and squamous cell metaplasia may be present, as well as mesenchymal cell accumulation and fibrosis. With progression of the condition, structural remodeling may occur, characterized by increased collagen content and scar tissue formation that narrows the airways and produces fixed airway obstruction (Figure 41-2).
Emphysema
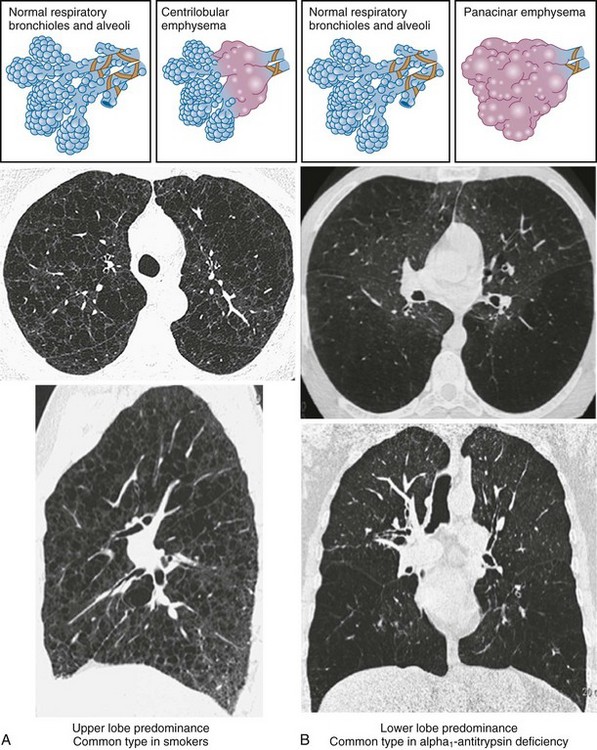
• Centrilobular (or centriacinar) emphysema, in which large air spaces are initially clustered around the terminal bronchiole (Figure 41-3, A).
• Panlobular (or panacinar) emphysema, where the large air spaces are distributed throughout the acinar unit (Figure 41-3, B).
Etiology
Risk Factors
Cigarette Smoking
In adulthood the effect of smoking on FEV1 decline is well known. In general there is a significant dose-response effect, with smokers having lower lung function the more and the longer they smoke. There is, however, considerable variation. Most longitudinal studies indicate that the decline in FEV1 in smokers ranges from 45 to 90 mL per year, in contrast to the normal 30 mL/yr (Figure 41-4). However, values vary considerably among individuals, and some experience significantly greater decline, at least temporarily, which may explain why COPD may seem to surface over a short period in the fifth and sixth decades of life. Some nonsmokers have impaired lung function, and 15% to 20% of COPD patients are lifelong nonsmokers. Conversely, some heavy smokers are able to maintain normal lung function, although the frequently quoted “15% to 20%” of smokers who are thought to develop clinically significant COPD is probably an underestimate. About 35% of smokers with normal lung function initially developed COPD during a 25-year follow-up in the Copenhagen City Heart Study.
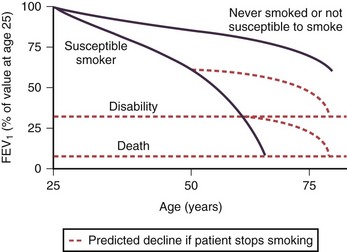
Pipe and cigar smokers have significantly greater morbidity and mortality from COPD than nonsmokers, although the risk is less than that from cigarettes. There is a trend to an increased relative risk of chronic airflow limitation from passive smoking, but the effect is not powerful enough to demonstrate clinical significance. Epidemiologic studies have associated cessation of smoking with a decrease in the prevalence of respiratory symptoms and improvement in the subsequent decline in FEV1 (Figure 41-4). The first effect on lung function after smoking cessation is a small increase of 50 to 100 mL in FEV1. There is some debate on whether decline in FEV1 after smoking cessation completely normalizes, although in general those who quit smoking continue to have an FEV1 decline slightly larger than in those who never smoked.
Host Factors
Genetic Factors
Chronic obstructive pulmonary disease is a prime example of a condition of gene-environment interaction. The observation of a familial association for an increased risk of airflow limitation in smoking siblings of subjects with severe COPD suggests a genetic component to this disease. Genetic linkage analysis has identified several sites in the genome that may contain susceptibility genes, such as chromosome 2q. Genetic association studies show that a number of candidate genes are associated with the development of COPD or with rapid decline in FEV1. However, the associations are not consistent in different populations (Box 41-2).
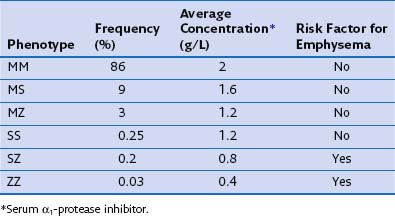
The most consistent association with COPD is alpha1-antitrypsin (α1-proteinase inhibitor) deficiency. Alpha1-antitrypsin is a glycoprotein that is the major inhibitor of serine proteases, including neutrophil elastase. More than 75 biochemical variants of α1-antitrypsin have been described relating to their electrophoretic properties, giving rise to the phase inhibitor (Pi) nomenclature (Table 41-1). The most common allele in all populations is PiM, and the most common genotype is PiMM, which occurs in 93% of the alleles in subjects of Northern European descent. PiMZ and PiMS are the next two most common genotypes and are associated with α1-proteinase inhibitor levels of 15% to 75% of the mean levels of PiMM subjects. Similar levels occur in the much less common PiSS type. The most important other type is PiSZ, in which basal levels are 35% to 50% of normal values. The threshold point for increased risk of emphysema is a level of about 80 mg/dL, which is about 30% of normal.
Table 41-1 Alpha1-Antitrypsin Phenotypes: Frequency in UK Population, Concentration, and Emphysema Risk