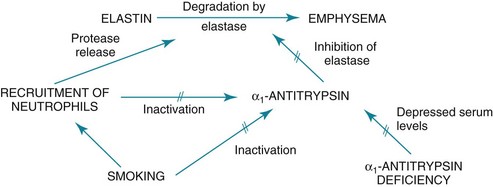
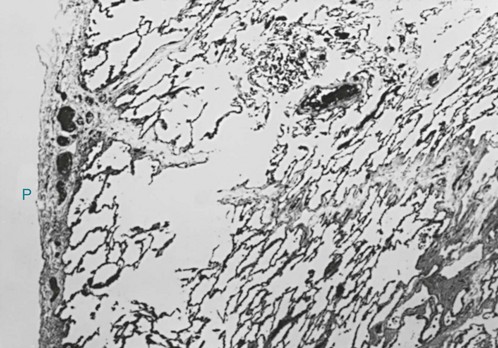
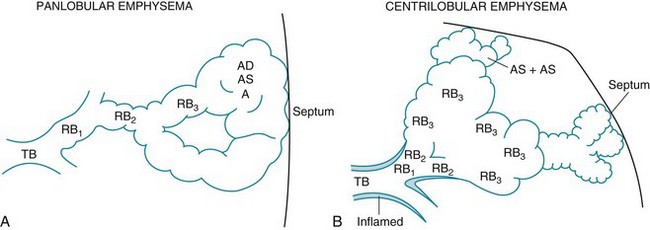
6 The term chronic obstructive pulmonary disease (COPD) refers to chronic disorders that disturb airflow, whether the most prominent process is within the airways or within the lung parenchyma. The two disorders generally included in this category are chronic bronchitis and emphysema. Although the pathophysiology of airflow obstruction is different in the two disorders, patients frequently have features of both, so it is appropriate to discuss them together. Asthma could logically also be in this category, but it is discussed in Chapter 5 because the term COPD, as commonly used, does not usually include bronchial asthma. In smokers, the balance between elastase and anti-elastase is thought to be disturbed in more than one way by cigarette smoke. First, an increased number of neutrophils can be found in the lungs of smokers, providing a source for increased amounts of neutrophil elastase. Second, evidence indicates that oxidants derived from cigarette smoke and inflammatory cells can oxidize a critical amino acid residue of α1-antitrypsin at or near the site where the protease inhibitor binds to elastase. Oxidation of this amino acid interferes with the inhibitory activity of α1-antitrypsin, again tipping the balance in favor of increased elastase activity. Hence, cigarette smoking may be a compound insult, increasing the amount of neutrophil elastase in the lung and decreasing the normal inhibitory mechanism that serves to limit uncontrolled elastin breakdown by the enzyme. This pathogenetic sequence hypothesized for the development of emphysema is summarized in Figure 6-1. Much of the pathology in chronic bronchitis relates to mucus and the mucus-secreting apparatus in the airways. Mucus-secreting glands and goblet cells are responsible for production of bronchial secretions, but the mucous glands are the more important source (see Chapter 4). In chronic bronchitis, enlargement (hypertrophy and hyperplasia) of the mucus-secreting glands has been objectively assessed by comparing the relative thickness of the mucous glands with the total thickness of the airway wall. This ratio, known as the Reid index, is increased in patients with chronic bronchitis. In general, the number of goblet cells in the airways is increased as well, and these particular cells are abundant in airways more peripheral than usual. These alterations in the mucus-secreting apparatus increase the quantity of airway mucus, and its composition is likely altered as well. In practice, the secretions found in patients often are thick and more viscous than usual. Bronchial walls demonstrate evidence of an inflammatory process, with cellular infiltration and variable degrees of fibrosis. In patients with severe chronic airflow obstruction, the most important process responsible for airflow obstruction is emphysema. As mentioned earlier, the pathology of emphysema is characterized by destruction of alveolar walls and enlargement of terminal air spaces (Fig. 6-2). Several types of emphysema have distinct pathologic features, primarily dependent on the distribution of the lesions. The most important types are panacinar (panlobular) emphysema and centriacinar (centrilobular) emphysema (Fig. 6-3). Panacinar emphysema is characterized by a relatively uniform involvement of the acinus, the region beyond the terminal bronchiole, including respiratory bronchioles, alveolar ducts, and alveolar sacs. Examination of a section of lung with panacinar emphysema shows that the damage in an involved area is relatively diffuse (Fig. 6-4). Typically the lower zones of the lung are more involved than the upper zones. Panacinar emphysema is the usual type of emphysema described in patients who have α1-antitrypsin deficiency, although the condition is not limited to this clinical setting.
Chronic Obstructive Pulmonary Disease
Etiology and Pathogenesis
Smoking
Pathology
Chronic Obstructive Pulmonary Disease