Figure 3-1.
Although Otto Loewi discovered chemical transmission of nerve impulses using acetylcholine [1], it was von Euler who discovered in 1946 that the true agent of sympathetic transmission is noradrenaline, also know as NE [2]. That led to the differentiation of adrenergic and cholinergic neurons and synapses. On left above is Otto Loewi’s experiment where he took fluid from one frog heart which had been stimulated, and applied it to another. The result was slowing of the second heart, which showed that synaptic signaling used chemical messengers. On the right above is von Euler’s description of the adrenergic nerve terminal based on his experiments [3] (Taken from his Nobel Award presentation on 12/1/1970).
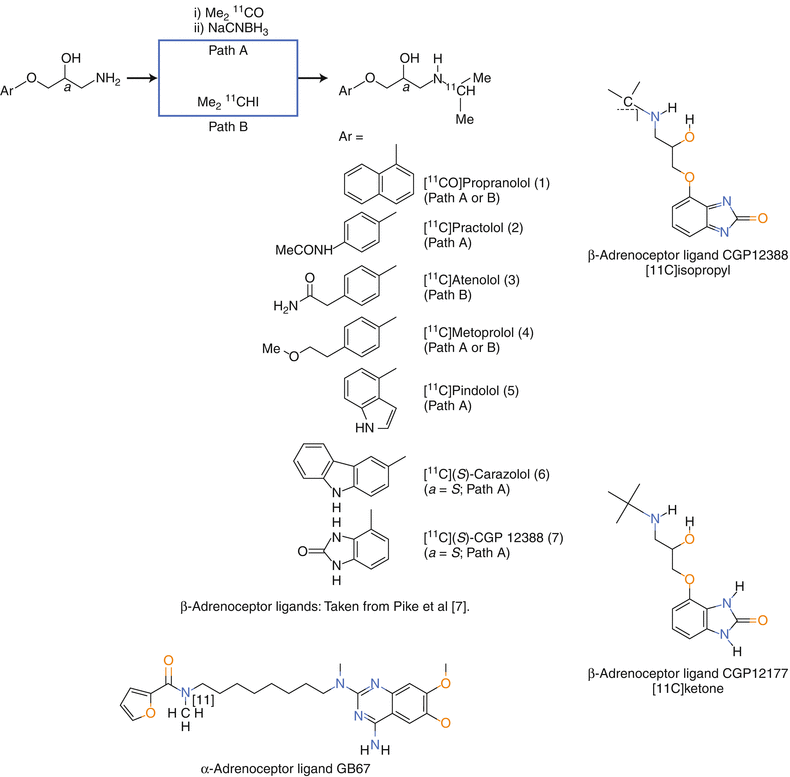
Figure 3-2.
Positron emission tomography (PET) imaging of receptors in the sympathetic and the parasympathetic systems of heart and lung has been carried out for three systems, the alpha and beta adrenoceptors (BAR) and the muscarinic acetylcholine receptors (mAChR). For the alpha-adrenoceptor, only [11C]GB67 (N2-[6-[(4-amino-6,7-dimethoxy-2-quinazolinyl)(methyl)amino]hexyl]-N2-[11C]methyl-2-furamide hydrochloride) has been developed. Its potential for application in patients needs to be assessed [4, 5]. The mAChR radioligand [11C]MQNB (N-[11C]methylquinuclidinyl benzilate) was the highest affinity radiotracer for the mAChR straightforward chemistry using the N-[11C]methylation approach [6]. The early BAR radioligands were based on the first drugs for that receptor, i.e., propranolol, practolol, atenolol, metoprolol, etc. [7] and upper left structures. In general, drugs seldom lead to ideal radiotracers given their weaker affinities and higher log P [8]. The most promising BAR radioligands are: [11C]CGP12177: S-(3′-t-butylamino-2′-hydroxypropoxy)-benzimidazol-2-[11C]one [9, 10] and [11C]CGP12388: (S)-4-(3-(2′-[11C]isopropylamino)-2-hydroxypropoxy)-2H-benzimidazol-2-one [11] and qualitatively in lung [11C]VC002: N-[11C]-methyl-piperidin-4-yl-2-cyclohexyl-2-hydroxy-2-phenylacetate [5]. [11C]-S-CGP 12177 had high specific uptake in the heart and lungs (both tissues have high density BAR). This was a success by the accepted technical design criteria, except for the challenging synthesis. A synthesis to introduce the C-11 in the last step led to [11C]CGP12388, also a high affinity radioligand [9–11]. However, the uptake was rapid with minimal efflux indicating that the concentration in the heart and lung was influenced by flow. Therefore an approach to obtain the Bmax using at least two contractions of radioligand was proposed based on the Scatchard equation, B/F = Bmax/Ki−B/Ki where the second term is considered negligible if the bound radioligand is <5 % of the Bmax [12]. In summary, for both the beta-adrenergic and the muscarinic systems, potent radioligands have been prepared and evaluated in patients. For both it has been possible to measure receptor densities quantitatively in human heart, but not by the single scan technique that is used routinely in the clinic (Taken from Pike et al. [7]).
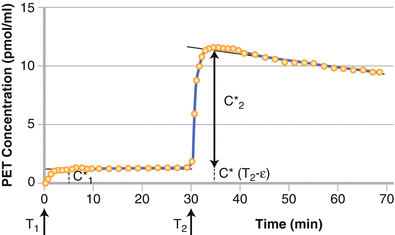
Figure 3-3.
The approach for the two injection/ two scan technique to estimate β-adrenergic receptor density using a graphic method. The experimental protocol includes two injections with doses D1* and D2* at times T1 and T2, respectively. The graphic method is based on measurement of 2 plateau levels at end of distribution phase (C1* and C2*). From the analytical data using the specific activity of 29,415 MBq/μmol with D1* = 3.09 nmol and D2* = 22.19 nmol and using a vascular fraction of 40 %, receptor concentration was estimated at 18.6 pmol/mL. As demonstrated, the clinical implementation of this requires acquisition of a time-activity curve and at least two images, one at the NCA level and one at ~50 % saturation. This approach is complicated and the target density (Bmax) is difficult to validate using ex vivo data and therefore only a few studies have been pursued by other groups in the routine clinic [13]. It appears that the initial biodistribution of beta adrenergic binding radioligands is heavily weighted by flow. In addition, the target density (Bmax) is not high compared to the affinity constant. The metrics derived from mathematical analysis using the two injection system using the most easily synthesized beta adrenoceptor ligand (CGP 12388) yielded Bmax values of 9.74 ± 1.80 nM and a Kd of 0.58 ± 0.22 nM. This yields a Bmax/Kd = 17, which a rule of thumb from in vitro studies of the maximum bound to free ratio to be expected in vivo, One other ramification of this ratio is that a high ratio will be beneficial as the field moves to imaging abnormal tissue that is smaller than twice the instrument resolution [27].
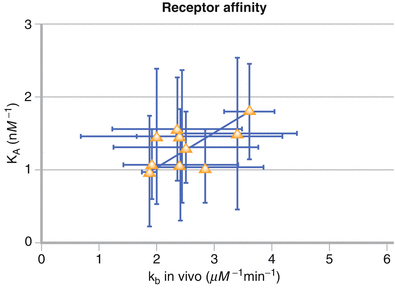
Figure 3-4.
Model parameter kb and an independent measurement of 99mTc-NGA-HBP affinity, KA, were highly correlated. The correlation was determined by weighted linear regression. Approaches to measuring receptor density for receptor binding radiotracers whose early distribution is heavily weighted by flow and the parent compound toxicity is low. The best example of this is the validation of in vivo receptor measurements via in vitro radioassay: Technetium-99m-Galactosyl-Neoglycoalbumin as Prototype Model [14]. The authors point out various stages of validation:
The ligand should not produce a physiologic effect
The localization in vivo should be receptor mediated
An alteration in biodistribution should reflect a change in disease or disease treatment
A pharmacokinetic method that allows quantitation of changes in receptor density should be developed and validated
Kinetic sensitivity of the analysis with respect to published changes in receptor density as a function of disease should be verified
Receptor density in tissue obtained from images should agree with that measured ex vivo
The key factor is sensitivity to changes in receptor density. Based on in vitro data developed from the original radioimmunoassays, a saturation of ~50 % will yield maximal change in binding as a function of receptor density changes.
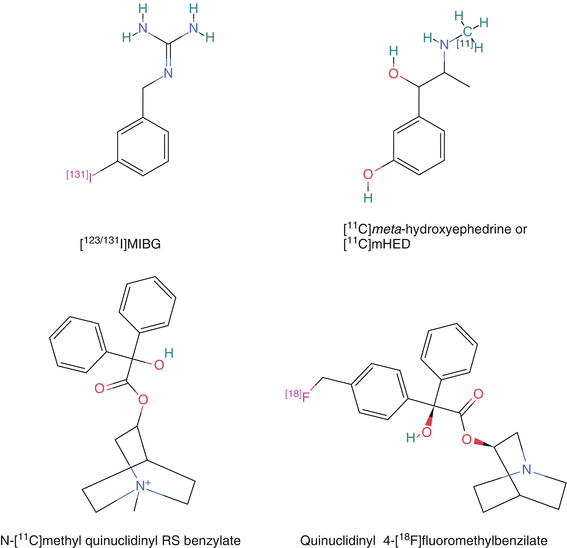
Figure 3-5.
Chemistry: Compared to N-methylation using C-11 methyl iodide or methyl triflate, preparing the precursor for radiofluorination or radioiodination is complicated. N-[11C]methyl quinuclidinyl benzoate was prepared using this method [16, 17]. The original method to produce radiolabeled QNB inserted the C-11 in the benzylic acid. This was achieved by carbonation of the benzophenone dianion followed by ester formation with 3-quinuclidinol [18–20]. Both were validated as specifically targeting the muscarinic receptor but the MQNB was used more often given the straightforward chemistry. The C-11 synthesis can also be complicated if the C-11 is not inserted in the final chemical step. For the two beta adrenoceptor ligands [11C]CGP12177 and [11C]CGP12388, the former is a complicated synthesis starting with C-11 phosgene followed by a multistep synthesis whereas the latter is a more straightforward radiolabeling of the isopropyl group via a reductive alkylation using [11C]acetone of the corresponding (S)-desisopropyl compound [9–11]. The complicated synthesis for [11C]CGP12177 prevented a detailed clinical study. Quinuclidinyl 4-[18F]fluoromethyl-benzilate was also developed and validated as binding to the muscarinic receptors and could be imaged in the monkey myocardium, but showed uptake in bone [21]. [123I]MIBG was originally synthesized by an exchange method using the non radioactive MIBG. Later, the trialkyl tin precursor was used to increase the specific activity. A procedure using a solid phase resin containing the covalently bound stannylbenzyl-guanidine precursor has the potential to produce the highest specific activity [22]. This is important given the potential side effects of carrier MIBG (Taken from PubChem [15]).
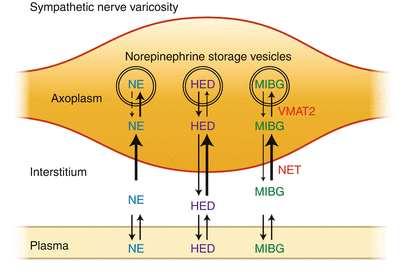
Figure 3-6.
Schematic illustration of the uptake of norepinephrine (NE), [11C]meta-hydroxyephedrine (HED), and meta[123I]iodobenzylguanidine (MIBG) into cardiac sympathetic nerve varicosities. After extraction from plasma, HED and MIBG are transported into sympathetic nerve terminal axons by the norepinephrine transporter (NET). Once in the neuronal axoplasm, they are subsequently transported into NE storage vesicles by the second isoform of the vesicular monoamine transporter (VMAT2). Because the radiotracers targeting receptors, enyzmes, and transporters of the sympathetic and parasympathetic nervous system are small molecules, the radionuclides used were chosen to introduce the minimal perturbation to the parent drug candidate from C-11, which replaced a stable carbon and therefore did not alter the biochemistry, to F-18 and I-123 that produced changes in the biochemistry. The C-11 radioligands had the biochemical properties of the parent compound, often a well-characterized drug candidate. The F-18 and I-123 analogs showed similarities in the biochemistry compared to the parent drug candidate, but validation studies were needed to elucidate the key biochemical parameters. This requires extensive effort using in vitro and in in vivo models. For example, the biochemistry of [18F]Fludeoxyglucose and [18F]Fluorothymidine are similar to that of deoxyglucose and thymidine, but there are key differences and the biochemistry of fluorothymidine is more unlike that of thymidine than fluorodeoxyglucose is from deoxyglucose (and glucose) [24] (Taken from Raffel [23]).
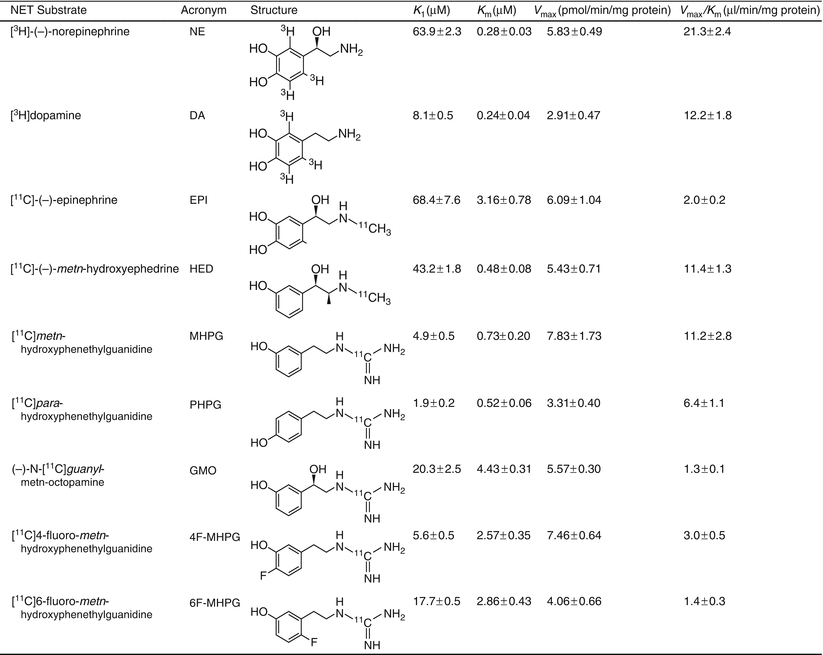
Figure 3-7.




NET substrates have been more completely validated for measuring the kinetics of various key biochemical steps. The goal is a diagnostic test per se, but also increasing the understanding of the underlying mechanisms of autonomic dysfunction and its contribution to the manifestation and progression of cardiac diseases. The search continues for a radioligand whose distribution is not primarily affected by flow or by endogenous NE, but rather sensitive to moderate levels of cardiac denervation. One recent attempt is to find a radiotracer that has a lower extraction than MIBG or HED. The new lead compound is 4-[18F]fluoro-m-hydroxyphenethyl-guanidine (4-[18F]F-MHPG) [26]. The compound was originally compared to NE and HED using the Michaelis-Menten enzyme analysis. The analysis in the current study was analyzed using a compartment model (net uptake Ki = 0.341 mL/min/g) and the Patlak analysis (Kp = 0.302 mL/min/g). The radio tracer, 4-[18 F]FMHPG, has a weaker Km with about the same maximum velocity, which was the design goal. It is worth noting that the Ki from Fig. 3.7 measures the inhibition of these ligands in the micromolar range. If the goal were to inhibit the transporter (as it is in the brain) then these affinity constants would have to be in the nanomolar range [28]. For substrates, the main concern is the competition with endogenous NE and therefore one design goal is to develop a substrate that binds with an affinity less than that of NE, which is 63.9 μM in this assay (Taken from Raffel et al. [25]).
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
