(Fig. 10.1). Elimination occurs from the central compartment and can be described by the elimination rate constant. By measuring plasma concentrations over a time period from the injection of the drug, it is possible to describe the concentration-versus-time profile for the drug (Fig. 10.2) (22). Distribution and elimination phases can be determined and a mathematical description (model) of the change in drug concentration versus time can be developed.
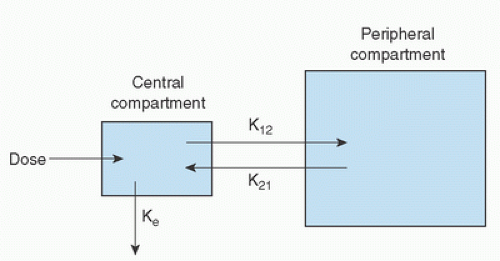 FIGURE 10.1. A two-compartment pharmacokinetic model illustrating the distribution of a drug within a central compartment (blood) and peripheral compartment (tissues), and its ultimate biotransformation and elimination from the body. K12 and K21 are first-order rate constants for transfer of drug between the peripheral and central compartments, whereas Ke is the elimination rate constant. (From Hall RI, Thomas BL, Hug CC Jr. Pharmacokinetics and pharmacodynamics during cardiac surgery and cardiopulmonary bypass. In: Mora CT, ed. Cardiopulmonary bypass: Principles and techniques of extracorporeal circulation. New York, NY: Springer-Verlag, 1995:56.) |
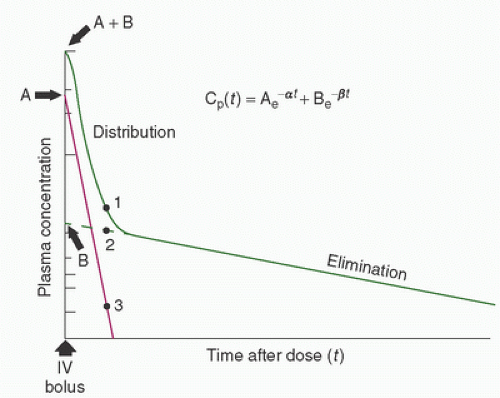 FIGURE 10.2. Plasma [log] concentration-versus-time curve for a hypothetical drug after a single intravenous dose. The curve (A + B) is the sum of the contributions from the rapid distribution (A) phase and the slow elimination (B) phase to the logarithmic decline in concentration after a bolus dose. The concentration at any time is given by the equation Cp(t) = Ae-at + Be-βt, where Cp(t) is the drug concentration in plasma at time t; A, constant determined from the Y-axis intercept (time = 0) of the distribution portion of the log concentration-versus-time curve, derived by subtracting the contribution of the (constant, first order) elimination phase of the curve; α, slope of the log concentration-versus-time curve of the distribution phase, derived by subtracting the contribution due to elimination; B, constant determined from the Y-axis intercept (time = 0) of the elimination phase of the log concentration-versus-time curve; β, slope of the log concentration-versus-time curve of the elimination phase. (From Hall RI, Thomas BL, Hug CC Jr. Pharmacokinetics and pharmacodynamics during cardiac surgery and cardiopulmonary bypass. In: Mora CT, ed. Cardiopulmonary bypass. Principles and techniques of extracorporeal circulation. New York: Springer-Verlag, 1995:56.) |
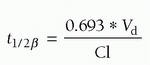
as drug administration continues, there is accumulation of drug in tissues over time, which increases with the duration of drug infusion. On termination of the infusion, offset of drug effect then depends on redistribution of the drug out of tissues (greater with longer infusions) back into the central compartment as well as the rate of elimination. Thus, when a drug has accumulated in peripheral tissues, reliance on the elimination half-time will not adequately predict termination of drug effect because it does not take into account the role of redistribution. This has led to the introduction of the term context-sensitive half-time as a better description of the phenomenon of increased duration of drug effect with increased drug infusion time (Fig. 10.3) (44).
TABLE 10.1. Definition of basic pharmacokinetic parameters | |||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||
of the population studied (e.g., young, healthy men versus elderly women with congestive heart failure [CHF]). Drug in the blood exists in several forms, including that which is free (active), bound to plasma proteins (e.g., albumin, and therefore subject to changes in plasma protein concentrations), or sequestered in red blood cells. CPB has the potential to alter all of these factors, which makes the description of pharmacokinetic parameters during CPB problematic.
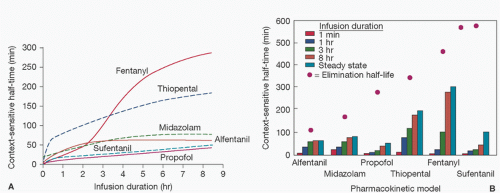 FIGURE 10.3. A: Context-sensitive half-times as a function of infusion duration for each pharmacokinetic model simulated. Solid and dashed lines are used to permit overlapping lines to be distinguished. B: Context-sensitive half-times (bars) redrawn from (A) for each pharmacokinetic model after terminating a 1-minute, 1-hour, 3-hour, 8-hour, or infinitely long (i.e., to steady state) computer-designed infusion designed to instantaneously achieve and maintain a target concentration shown relative to the elimination half-life (dots) computed for each model. (From Hughes MA, Glass PSA, Jacobs Jr. Context-sensitive half-time in multicompartment pharmacokinetic models for intravenous anesthetic drugs. Anesthesiology 1992;76:336.) |
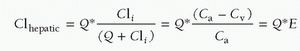
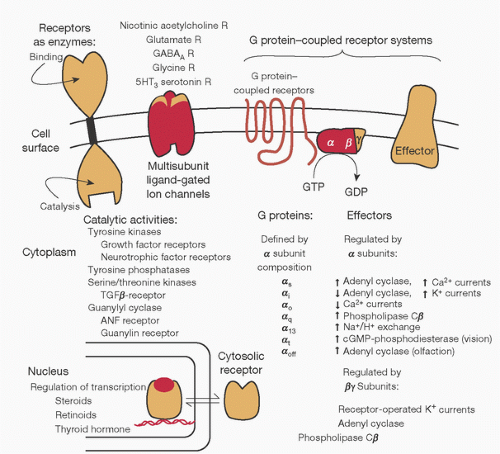 FIGURE 10.4. Structural motifs of physiologic receptors and their relation to signaling pathways. Schematic diagram of the diversity of mechanisms for control of cell function by receptors for endogenous agents acting through the cell surface or in the nucleus. (From Ross EM. Pharmacodynamics. Mechanisms of drug action and the relationship between drug concentration and effect. In: Hardman JG, Limbird LE, eds. Goodman and Gilman’s the pharmacological basis of therapeutics. 9th ed. Montreal, QC: McGraw-Hill, 1996:32.) |
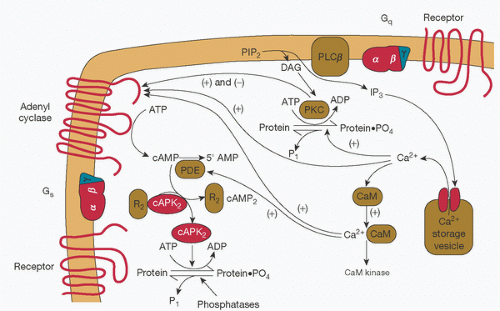 FIGURE 10.5. Interactions between the second messengers cyclic adenosine monophosphate (cAMP) and Ca2+. Generation of second messengers cAMP and Ca2+ permits distribution of cell-surface regulatory input into the cell interior, amplification of the initial signal, and opportunities for synergistic or antagonistic regulation of other signaling pathways. PIP2, phosphatidylinositol 4,5-biophosphate; DAG, diacylglycerol; IP3, 1,4,5-inositol triphosphate; CaM, calmodulin; R2, regulatory subunits of cyclic AMP-dependent protein kinase, which bind cyclic AMP; cAPK2, catalytic subunits of cyclic AMP-dependent protein kinase; PKC, protein kinase C, activated by DAG and Ca2+. (From Ross EM. Pharmacodynamics. Mechanisms of drug action and the relation between drug concentration and effect. In: Hardman JG, Limbird LE, eds. Goodman and Gilman’s the pharmacological basis of therapeutics. 9th ed. Montreal, QC: McGraw-Hill, 1996:34.) |
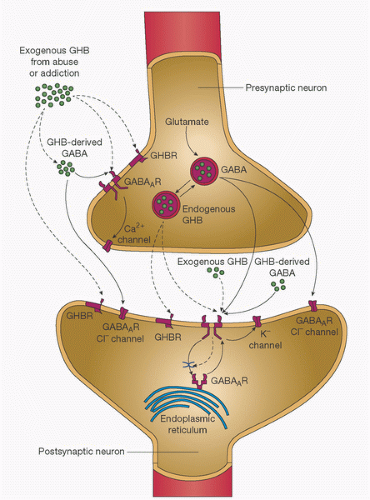 FIGURE 10.6. Synthesis and release of γ-hydroxybutyric acid (GHB) and γ-aminobutyric acid (GABA) at synapses—an example of a ligand-gated ion channel. The diagram shows the presynaptic and postsynaptic effects of endogenously released GHB (as indicated by dashed arrows) and (GABA) (as indicated by solid arrows) and the effects of exogenously administered GHB, as in abuse and addiction. GABA is synthesized from glutamate in inhibitory neurons and in turn gives rise to GHB. Both GHB and GABA are released upon depolarization of the GABA-releasing (GABAergic) presynaptic neuron. GABA, in forms that are either endogenous or derived from exogenously administered GHB, acts on GABAA and GABAB receptors (GABAAR and GABABR, respectively). GABAA receptors are ionotropic and, when activated by GABA, cause fast postsynaptic inhibition by the efflux of chloride ions (Cl-). GABAB receptors are metabotropic and, when activated by either GABA or high concentrations of GHB, induce slow postsynaptic inhibition by activating outward potassium (K+) currents. Presynaptic GABAB autoreceptors—when activated by GHB, GABA, or both—reduce the release of GABA by suppressing the influx of calcium (Ca2+). Both endogenous and exogenous forms of GHB have a dual action on the GHB receptor (GHBR) and the GABAB receptor. GHB that binds with high affinity to the presynaptic GHB receptor decreases the release of GABA; GHB that binds to a low-affinity site on the GABAB receptor increases activation of cell-surface receptors by inhibiting constitutive and agonist-induced endocytosis. The result is enhancement of GHB function mediated by GABAB receptors, with a greater effect on presynaptic inhibition than on postsynaptic inhibition. (From Snead OC III, Gibson KM. γ-Hydroxybutyric acid. N Engl J Med 2005;352:26;2724.) |
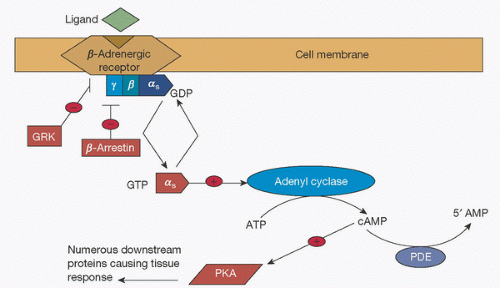 FIGURE 10.7. G-protein-coupled receptors. Schematic of signal transduction cascade for receptors coupling to G-protein [α]s subunit. ATP, adenosine triphosphate; cAMP, cyclic adenosine monophosphate; PDE, phospho-diesterase; PKA, protein kinase A; GRK, G-protein receptor kinase; GDP, guanosine diphosphate; GTP, guanosine triphosphate. (From Johnson JA, Lima JJ. Drug receptor polymorphisms and pharmacogenetics: current status and challenges. Pharmacogenetics 2003;13(9):531.) |
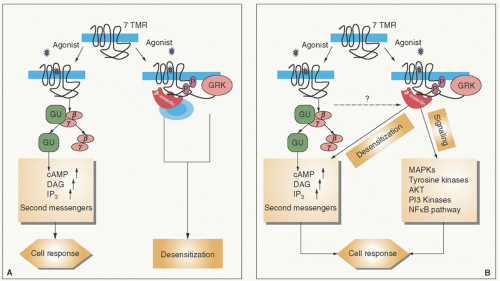 FIGURE 10.8. Signal transduction by seven transmembrane receptors. A: Classical paradigm. The active form of the receptor (R*) stimulates heterotrimeric G-proteins and is rapidly phosphorylated by GRKs, which leads to β-arrestin recruitment. The receptor is thereby desensitized, and the signaling is stalled. B: New paradigm. β-Arrestins not only mediate desensitization of G-protein-signaling but also act as signal transducers themselves. TMR, transmembrane receptor; cAMP, cyclic adenosine monophosphate; DAG, diacylglycerol; IP3, 1,4,5-inositol triphosphate; Gα,γ,β, trimeric G-protein subunits; GRK, G-protein-coupled receptor kinases; MAPKs, mitogen-activated protein kinases; AKT, protein kinase B pathway; PI3, phosphatidylinositol-3-kinase pathway. (From Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by β-arrestins. Science 2005;308:513.) |
TABLE 10.2. Examples of G-protein-coupled receptors and the intracellular second messengers they generate | ||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||
regulation of several different Ca2+-specific channels in the plasma membrane and by its release from intracellular storage sites (Fig. 10.12) (69,86,89). Ca2+-dependent ion channels are opened by electrical depolarization, phosphorylation by a cAMP-dependent protein kinase, and by Gs, K+, and Ca2+ itself (89). Opening of the channel may be inhibited by other proteins (e.g., Gi).
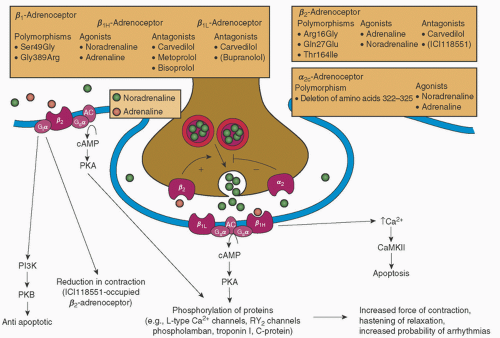 FIGURE 10.9. Features of the cardiac sympathetic nervous system. (—)-Noradrenaline released from sympathetic nerve terminals is complemented by circulating (—)-adrenaline. The release of (—)-noradrenaline is modulated by facilitatory, prejunctional β2-adrenoceptors and autoinhibitory, prejunctional α2-adrenoceptors. A deletion polymorphism of the autoinhibitory α2c-adrenoceptor reduces its function and leads to heightened noradrenaline release from prejunctional nerve terminals. In heart failure, the activity of the sympathetic nervous system increases, with a consequent increase in the plasma concentration of (—)-noradrenaline. (—)-Noradrenaline activates postjunctional β1-adrenoceptors that couple to the Gsα-cAMP pathway. Gsα activates adenyl cyclase (AC), which catalyzes the formation of cAMP. cAMP, in turn, activates cAMP-dependent protein kinase (PKA). PKA phosphorylates several proteins that contribute to increased force of contraction and hastening of relaxation. There are two forms of β1-adrenoceptors, β1H and β1L. In the human heart, β2-adrenoceptors also couple to the Gsα-cAMP pathway. Recently, it has been demonstrated that β1- and β2-adrenoceptors couple to additional signaling pathways that are of interest in heart disease. These include β2-adrenoceptor stimulation of Giα signaling pathways. Agonist-activated β2-adrenoceptor-Giα signaling has putative antiapoptotic effects through phosphatidylinositol 3-kinase (PI3K)-protein kinase B (PKB) signaling. The β2-adrenoceptor antagonist ICI118551 reduces human ventricular myocyte maximal shortening and contraction duration. β1-Adrenoceptor-mediated increase in Ca2+ stimulates Ca2+/calmodulin-dependent protein kinase II (CaMKII) proapoptotic signaling in animals. Antagonists given in parentheses are not used in the management of heart failure. RY2 channels, ryanodine RY2 receptor channels. (From Molenaar P, Parsonage WA. Fundamental considerations of β-adrenoceptor subtypes in human heart failure. Trends in Pharmacol Sci 2005;26:369.) |
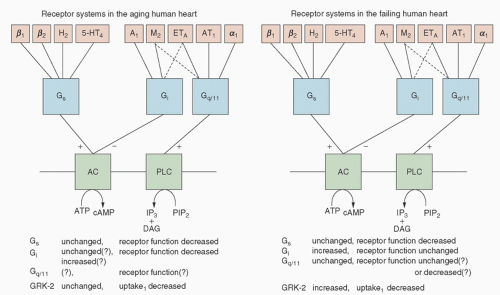 FIGURE 10.10. Receptor systems and their signal transduction mechanisms in the aging (left) and failing (right) human heart. β1, β2, α1 Stands for β1-, β2-, and α1-adrenoceptors, respectively; H2, histamine H2-receptors; 5-HT4, serotonin 5-HT4-receptors; M2, muscarinic M2-receptors; A1, adenosine A1-receptors; ETA, endothelin ETA-receptors; AT1, angiotensin II AT1-receptors; Gs, stimulatory G-protein; Gi, inhibitory G-protein; Gq/11, the G-protein that couples ET-, AT-receptors and α1-adrenoceptors to phospholipase C (PLC); AC, adenyl cyclase; PIP2, phosphatidylinositol 4,5-bisphosphate; DAG, diacylglycerol; IP3, inositol trisphosphate; GRK, G-protein-coupled receptor kinase; uptake1, noradrenaline reuptake transporter (+, activation; -, inhibition). (From Brodde OE, Leineweber K. Autonomic receptor systems in the failing and aging human heart: similarities and differences. Eur J Pharmacol 2004;500:168.) |
An immediate reduction in concentrations of circulating proteins such as albumin and a1-acid glycoprotein (α1AGP). This has implications for protein binding of drugs resulting from alteration in the ratio of bound drug to free drug in the circulation (5,6,7,8,9,10,95,96,97,98,99,100,101,102,103).
An immediate reduction in red blood cell concentration, which has implications for compounds that are sequestered to a significant degree in red blood cells (3,8,104,105).
An immediate reduction in the amount of free drug in the circulation at the initiation of CPB. This will reduce the amount of drug available for interaction with the receptor, with the potential for adverse events, for example, lightening of the level of anesthesia (5,6,7,36,106).
Alteration in organ blood flow, which may affect drug distribution and clearance (107).
lipid solubility relative to the volume of the CPB prime. Thus, following intravenous administration, the tissues serve as a reservoir that sequesters the drug. At the onset of CPB, when plasma concentrations fall due to hemodilution, the drug moves down its concentration gradient from the tissue stores to plasma. As protein concentrations fall, the free drug concentration increases to reestablish an equilibrium based on its solubility in plasma (8,11,101). Because free drug concentration is responsible for drug effect (109,110), the increase in free drug concentration from altered protein binding may explain the increased pharmacodynamic effect observed during and after CPB (9,10,111).
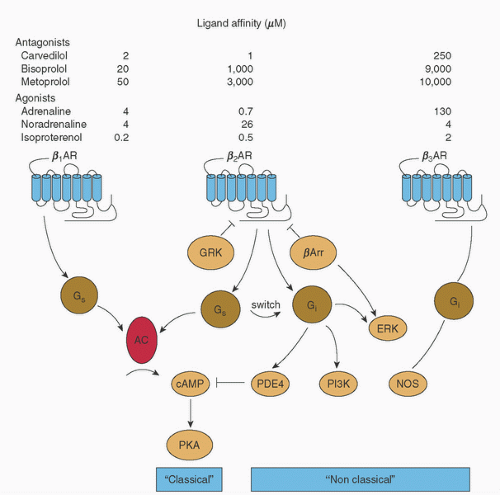 FIGURE 10.11. Agonist activation and coupling/signaling properties of β-adrenergic receptor subtypes. GRK, G-protein-coupled receptor kinase; βArr, β-arrestin; PDE, phosphodiesterase; PI3K, phosphatidylinositol 3-kinase; AC, adenyl cyclase; Gs, stimulatory G-protein; Gi, inhibitory G-protein; cAMP, cyclic adenosine monophosphate; PKA, protein kinase A; NOS, nitric oxide synthase; ERK, extracellular signal-regulated kinases. (From Lohse MJ, Engelhardt S, Eschenhagen T. What is the role of β-adrenergic signaling in heart failure. Circ Res 2003;93:897.) |
Peripheral vasoconstriction may decrease absorption of drugs administered other than by the intravenous route (119).
Fluid extravasation may alter drug distribution from central to peripheral compartments (i.e., changes in the volume of distribution, Vd) (94,120).
Vasoconstriction may reduce the rate of reuptake of drug from peripheral tissues to the central compartment (120,121).
Temperature-induced reductions in enzyme-mediated biotransformation may decrease clearance of drugs and increase their elimination half-time (Fig. 10.16) (120,121,122,123,124,125,126,127,128,129,130,131).
Changes in organ perfusion may produce altered renal drug excretion as a result of decreased renal perfusion, glomerular filtration rate, and tubular secretion (132). However, studies comparing renal function during normothermic versus hypothermic bypass have not demonstrated any clinically important differences (117,130,133,134).
Hypothermia-induced increases in drug solubility in blood of volatile anesthetics (135).
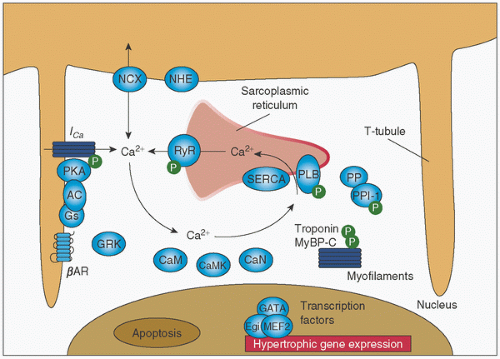 FIGURE 10.12. Calcium cycling in cardiac myocytes and regulation by protein kinase A (PKA). AC, adenyl cyclase; RyR, ryanodine receptor; PLB, phospholamban; SERCA, sarcoplasmic reticulum calcium ATPase; CaM, calmodulin; CaMK, calmodulin-dependent kinase; CaN, calcineurin; GRK, G-protein-coupled receptor kinase; NCX, sodium-calcium exchanger; NHE, sodium-proton exchanger; PP, protein phosphatase; Gs, stimulatory G-protein; BAR, β-adrenergic receptor; PPI-1, protein phosphatase inhibitor-1; MyBPC-C, myosin binding protein C, slow type; P, phosphorylation. (From Lohse MJ, Engelhardt S, Eschenhagen T. What is the role of β-adrenergic signaling in heart failure. Circ Res 2003;93:897.) |
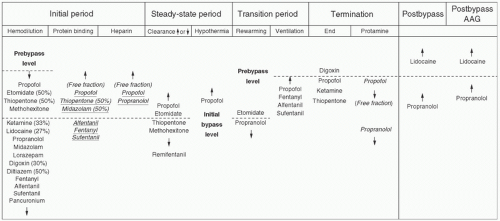 FIGURE 10.13. Plasma drug levels and factors affecting their concentration during cardiopulmonary bypass. All drug names indicate the level of plasma concentration unless the column denotes plasma free fraction change (italics and underlines). AAG increases after surgery. AAG, α1-acid glycoprotein. (From Mets B. The pharmacokinetics of anesthetic drugs and adjuvants during cardiopulmonary bypass. Acta Anaesthesiol Scand 2000;44:264.) |
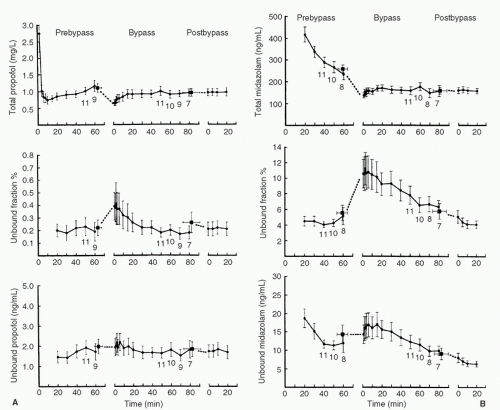 FIGURE 10.14. A: Total plasma concentration, unbound fraction, and unbound plasma concentration for propofol as a function of time for prebypass, bypass, and post-bypass periods. Each data point represents n = 12 (mean ± SEM). Numbers adjacent to the data points indicate n, where n is less than 12. The square data point to the left of t = 0 of the bypass period in each graph represents the mean of the final prebypass samples (mean ± SEM) plotted at the time (mean ± SEM) they occurred. The square data point to the left of the t = 0 of the post-bypass period in each graph represents the mean of the final bypass samples (mean ± SEM) plotted at the time (mean ± SEM) they occurred. Total plasma concentrations fall at the initiation of CPB, with little change in free drug concentrations, leading to an increase in the free fraction. B: Total plasma concentration, unbound fraction, and unbound plasma concentration for midazolam as a function of time for prebypass, bypass, and post-bypass periods. Each data point represents n = 12 (mean ± SEM). Numbers adjacent to the data points indicate n, where n is less than 12. The square data point to the left of the E = 0 of the bypass period in each graph represents the mean of the final prebypass samples (mean ± SEM) plotted at the time (mean ± SEM) they occurred. The square data point to the left of t = 0 of the post-bypass time period in each graph represents the mean of the final bypass samples (mean ± SEM) plotted at the time (mean ± SEM) they occurred. Total plasma concentrations fall at initiation of CPB, with little change in free drug concentrations, leading to an increase in free fraction. (From Dawson PJ, Bjorksten AR, Blake DW, et al. The effects of cardiopulmonary bypass on total and unbound plasma concentrations of propofol and midazolam. J Cardiothorac Vasc Anesth 1997;11:559.) |
esmolol, remifentanil, clevidipine, atracurium, and cis-atracurium) (123,124,125,127). The net result is a prolonged drug effect that requires dosage reductions.
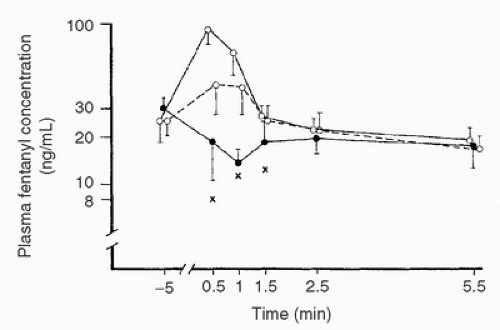 FIGURE 10.15. Plasma fentanyl concentrations (mean ± SD) in patients connected to cardiopulmonary bypass (CPB) circuits with primes containing no fentanyl (•—•) or containing a calculated fentanyl concentration of 140 (°—°) or 280 (°——°) ng/mL. X, lowest drug concentration measured at each stage during the first 1.5 minutes of CPB in patients not receiving fentanyl in their prime. NB: Regardless of whether the prime is supplemented or not, no difference exists in fentanyl concentrations within 2.5 minutes. (From Hynynen M. Binding of fentanyl and alfentanil to the extracorporeal circuit. Acta Anaesthesiol Scand 1987;31:708.) |
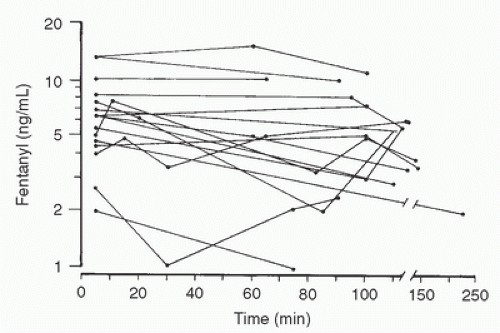 FIGURE 10.16. Fentanyl plasma concentrations in 18 children during profound hypothermia (18°C-25°C). Time zero is the initiation of cardiopulmonary bypass. Total plasma fentanyl levels remain essentially unchanged. (From Koren G, Barker C, Goresky G, et al. The influence of hypothermia on the disposition of fentanyl—human and animal studies. Eur J Clin Pharmacol 1987;32:374.) |
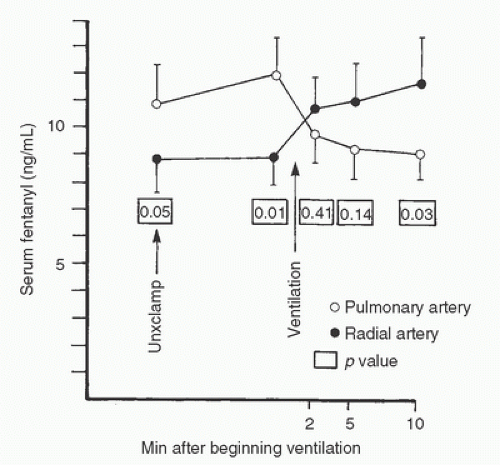 FIGURE 10.17. Mean fentanyl levels in seven cardiac surgery patients as ventilation and perfusion to the lung are resumed near the end of cardiopulmonary bypass. Systemic fentanyl concentrations rise with ventilation, whereas levels in the pulmonary artery fall, suggesting washout of fentanyl sequestered in the lungs during CPB. Unclamp, removal of aortic crossclamp. (Bently JB, Canahan TJ III, Cork RC. Fentanyl sequestration in lungs during cardiopulmonary bypass. Clin Pharmacol Ther 1983;34:705.) |
in whom postoperative lung concentrations of levofloxacin administered in the intensive care unit (ICU) were lower among those undergoing CPB versus a group not undergoing CPB. This finding was attributed to a higher degree of atelectasis (and hence altered tissue distribution) in postoperative CPB patients (151).
PLP group was associated with an increase in blood pressure and Bispectral Index (BIS) scores (BIS—a measure of anesthetic level), indicative of lightening of anesthesia, a finding which was not observed in the PMP group.
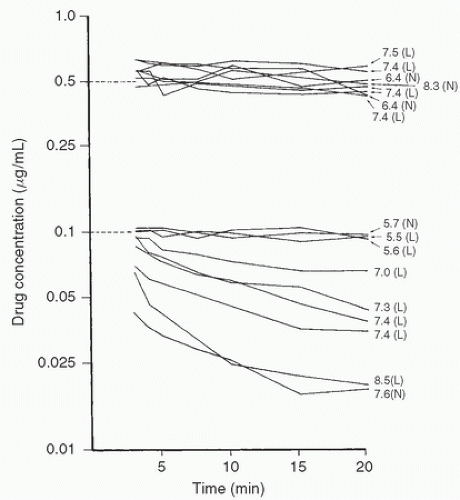 FIGURE 10.18. Changes in the concentrations of alfentanil (top) and fentanyl (bottom) in extracorporeal circuit prime with time (shown on the same logarithmic scale—vertical axis). Dotted lines represent predicted concentrations. Numbers on the right side are pH values of each priming solution. L, low temperature (24.4°C-25.70°C); N, normothermic (34.1°C-37.0°C); NB: The differences in binding to the cardiopulmonary bypass apparatus occur as a result of dissimilarities in the ionization of the two drugs with changes in pH. (From Skacel M, Knott C, Reynolds F, et al. Extracorporeal circuit sequestration of fentanyl and alfentanil. Br J Anaesth 1986;58:948.) |
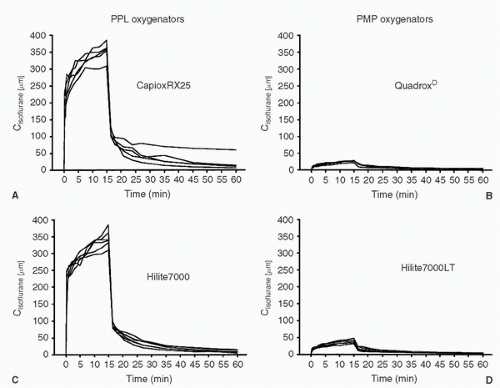 FIGURE 10.19. Isoflurane blood concentrations (Cisoflurane [µm]) for the uptake and elimination sequence in the four oxygenator groups. Each line represents a single patient. During hypothermic cardiopulmonary bypass, isoflurane 1% was administered to each patient. A: CapioxRX25; B: Hilite7000; C: QuadroxD; D: Hilite7000LT. (From Wiesenack C, et al. In vivo uptake and elimination of Isoflurane by different membrane oxygenators during cardiopulmonary bypass. Anesthesiology 2002;97:133-138.) |
theory the use of a cell saver device might have an effect on clearance of protein-bound drugs, this has not been demonstrated to be a clinical concern to date (197).
TABLE 10.3. Factors influencing movement across a membrane | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
slower return of drug from the effect site to the central compartment (a pharmacokinetic difference) (216). In contrast, a true difference in CNS sensitivity to opioids (fentanyl, alfentanil, and remifentanil) (217,218) and volatile agents (219) has been described in the elderly. For the elderly patient undergoing CPB, a reduction in dose to limit the high free drug concentrations and the potential for prolonged or toxic drug effects therefore seems prudent.
TABLE 10.4. Muscle relaxants and hypothermic cardiopulmonary bypass | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
of excitatory amino acid receptors in the spinal cord triggers intracellular events leading to Ca2+ release and phosphokinase C (PKC) activation. PKC is associated with a number of neuronal changes, including development of hyperalgesia (290). As tolerance to morphine administration shares many of the same pathways (290), studies in animals and humans have also implicated these pathways in the development of hyperalgesia (so-called opioid-induced hyperalgesia [OIH]) (291,293). Whether hyperalgesia develops in the perioperative period is debated, but recent meta-analysis concluded that, at least for the administration of remifentanil, it does (294). Representative studies, in patients undergoing cardiac surgery, include that of Richebe and colleagues (295) who examined the incidence of hyperalgesia following remifentanil administration by CACI with a target concentration of 7 ng/mL as compared to a group of patients receiving a zero-order infusion of 0.3 µg/kg/min. Use of the targeted infusion led to a reduction in total remifentanil dose and a reduction in hyperalgesia. In a randomized study of 40 patients undergoing coronary artery bypass graft (CABG) surgery comparing sufentanil targeted to a plasma concentration of 0.4 or 0.8 ng/mL, it was observed that the higher-dose sufentanil group had increased postoperative analgesic requirements, but assessments for hyperalgesia indicated that, while present and diminishing over time, the degree of hyperalgesia was not different between the two groups (296). In contrast, in a randomized study of 90 patients undergoing cardiac surgery and receiving either sufentanil 0.3 µg/kg/min intraoperatively or placebo (in addition to a sufentanil/propofol-based anesthetic), no difference in postoperative analgesic requirements was observed. Although the authors concluded that hyperalgesia did not occur (297), it must be acknowledged that it was not specifically tested for as in the studies of Fechner et al. (296) and Richebe et al. (295).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


