Fig. 39.1
Mechanism of action of methyldopa involving conversion to alpha-methylnorepinephrine and therein stimulation of central α2-adrenoreceptors and a reduction in sympathetic outflow
Table 39.1
Pharmacokinetics of centrally acting drugs
Drug | Volume of distribution (L/kg) | Absorption (%) | T max (h) | Half-life (h) | Plasma protein binding (%) |
|---|---|---|---|---|---|
α-Methyldopa | 0.6 | 25 | 2.0 | 1.7 | <15 |
Clonidine | 2.0 | 75–100 | 1.5–2.0 | 6–15 | 20–40 |
Guanabenz | 7.4–13.4 | 75 | 2–5 | 6–14 | 90 |
Guanfacine | 6.3 | >90 | 1.5–4.0 | 17 | 70 |
Moxonidinea | 3.0 | 80–90 | 0.5–3.0 | 2–3 | 5.8–7.9 |
Rilmenidinea | 315–325 L | 100 | 1.7 | 8.5 | 10–11 |
Table 39.2
Renal and pregnancy considerations with centrally acting drugs
Drug | Renal eliminationa (%) | Dialyzability | Dose adjustment in renal failure | FDA pregnancy category |
|---|---|---|---|---|
α-Methyldopa | 70 | Yes | Yes | C |
Clonidine | 58 | Yes, but limited | No | C |
Guanabenz | <5 | No | No | Cb |
Guanfacine | 50 | Limited | No | Bc |
Moxonidined | 50–75 | Not known | Yes | Not classified |
Rilmenidined | 52–93 | Yes | Yes | Not classified |
The onset of action varies among the compounds in this class with clonidine showing measurable activity within 15–30 min of intake. These compounds typically have a sizeable volume of distribution, which, in part, relates to their compartmentalization in the brain. The plasma half-life for compounds in this class can vary substantially from their pharmacodynamic half-life. This observation relates to receptor affinity and depot effects in deep tissue compartments. Finally, moxonidine and rilmenidine, compounds that are not available in the United States, are extensively renally cleared, which requires that they be judiciously dose-adjusted in the patient with reduced renal function [8, 9] (Table 39.2).
39.2.2 Pharmacodynamics
Centrally acting antihypertensive medications stimulate central vasomotor adrenergic receptors (e.g., nuclei tractus solitarii) and in so doing inhibit central sympathetic outflow to both the heart and the vasculature in multiple peripheral vascular beds. Plasma catecholamine levels fall with centrally acting therapy, which may relate, in part, to stimulation of peripherally situated presynaptic α2-receptors [10]. Clonidine stimulates both α2-receptors and imidazoline (I1) receptors as the basis for its peripheral sympathoinhibition [11]. Guanfacine is considered a more selective alpha2-receptor agonist than clonidine. Unlike clonidine, guanfacine does not have any measurable effect on dopamine turnover (Fig. 39.2).
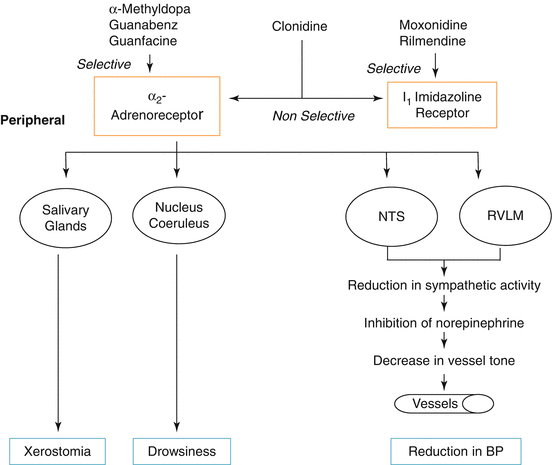
Fig. 39.2
Centrally acting antihypertensive medications working at either the α2-adrenoreceptor or the I1 imidazoline receptor. Xerostomia and sedation are mainly α2-adrenoreceptor mediated and are less common side effects with the I1 imidazoline receptor antagonists. NTS nucleus tractus solitarius, RVLM rostral ventrolateral medulla
The physiologic effects of withdrawal of SNS tone include similar and balanced falls in peripheral vascular resistance and systolic/diastolic BP. The reduction in peripheral resistance that is seen with centrally acting agents is maintained throughout long-term treatment. Despite the vasodilator action of drugs in this class, reflex tachycardia does not occur, and, in point of fact, heart rate may be dose-dependently reduced in the order of 5 % during treatment. Patients with renal insufficiency and clinical sinus node dysfunction and those who had developed bradycardia while taking other sympatholytic agents or who were currently receiving another sympatholytic drug are prone to greater heart rate reductions with clonidine. Cardiac output and renal blood flow typically go unaffected with drugs in this class [12]. Also, during exercise, these compounds still reduce peripheral vascular resistance, implying that exercise-related changes in SNS activity are prevented [13]. Centrally acting agents also reduce plasma renin activity and plasma aldosterone and with long-term treatment, left ventricular hypertrophy will regress [14]. Agents in this class tend to dose-dependently foster salt and water (H2O) retention and, as a result, their effectiveness may wane in the occasional patient over time [12]. This pseudotolerance to a BP-lowering effect can be undone with the addition of diuretic therapy.
39.3 Indications/Contraindications and Objectives of Therapy
39.3.1 Indications
The BP-lowering effect of both central α2-receptor and I1-receptor agonists is well established. Clonidine, the most commonly used drug in this class, was shown in the Veterans Affairs Cooperative Study to be an effective antihypertensive agent particularly in whites and older blacks [3]. Centrally acting compounds compare favorably in effectiveness to several first-line antihypertensive drug classes – such as thiazide diuretics (Chap. 38), angiotensin-converting enzyme (ACE) inhibitors (Chap. 36), and calcium channel blockers (CCBs) (Chap. 37). These compounds however probably find greater use as add-on therapies when sympathetic activation is an ongoing issue, either primary or secondary in nature. The latter might be the case when reflex sympathetic activation as occurs with vasodilators, such as hydralazine and minoxidil, needs to be checked (Chap. 40).
Centrally acting compounds are particularly useful in individuals whose hypertension has a significant anxiety component and is inherently labile. Drugs in this class can be used safely in patients with diabetes mellitus without any deterioration in glycemic control. Patients with pulmonary diseases, such as asthma, also tolerate these compounds even as they can produce xerostomia [15]. Clonidine is often used in the perioperative setting because it lessens sympathetically driven BP increases while also providing both an anesthetic- and analgesic-sparing effect [16].
39.3.2 Drug Differentiation and Mode of Delivery Considerations
All five drugs contained in the centrally acting medication class reduce BP similarly if equivalent doses are used although there are differences in the frequency of dosing between class members (Table 39.3). Clonidine and α-methyldopa are the only two drugs in this class, which are available in a parenteral form. Clonidine is the only compound in this class that is offered in a transdermal delivery system [17]. Differences in the onset and duration of action are two features that distinguish compounds in this class. Clonidine has the quickest onset of action and in most instances the shortest duration of effect. The half-life of drugs in this class widely varies and oftentimes does not predict the duration of effect per se [9, 11].
Table 39.3
Dosing considerations with centrally acting antihypertensive agents
Available compounds |
α-Methyldopa, clonidine, guanfacine, guanabenz, moxonidine, rilmenidine |
Dosing considerations |
α-Methyldopa – Oral α-methyldopa is initially given two to three times daily in a dose of 250 mg. Individual maintenance doses are in the range of 0.5–2.0 g/day in two to three divided doses |
Clonidine – Oral clonidine is best given two to three times daily. Starting doses are 0.1 mg two to three times daily with dose increase up to as high as 0.6 mg two to three times daily. A small dose of clonidine (0.1–0.2 mg twice daily) augments the BP-lowering effect of most other agents and can be reliably used as add-on therapy. Clonidine is a short-acting compound so patients with excessive sympathetic activity can have a short-lived response to it. If given together with a β-blocker, rebound hypertension is more common when clonidine is abruptly stopped and β-blocker therapy is continued. In some countries, transdermal clonidine is available. The transdermal systems dose range allows for release of 0.1–0.3 mg/24 h. There is a 1–2 days delay in the onset of action after initial patch application with transdermal clonidine, making it inappropriate for the management of hypertensive emergencies; conversely, removal of the transdermal delivery system for clonidine does not immediately eliminate drug effect. Clonidine can also be used intravenously for perioperative hypertension with a dose of 0.15 mg given two to four times daily. The side effects can also occur at lower doses |
Guanfacine – The initial response to guanfacine is delayed compared to clonidine but its longer duration of action allows it to be effectively dosed in a range of 1–3 mg given once or twice daily in a split dose. Withdrawal phenomena are significantly less pronounced than observed with clonidine, which may relate to its longer duration of action. Adverse effects increase significantly with doses in excess of 1 mg/day |
Guanabenz – Guanabenz is given by mouth as the acetate, but doses are usually expressed in terms of the base. Guanabenz acetate 5 mg is equivalent to about 4 mg of guanabenz. In hypertension, the usual dose is 4 mg twice daily initially; the daily dose may be increased by amounts of 4–8 mg every 1–2 weeks according to response. Doses of up to 32 mg twice daily have been used |
Moxonidine – 0.2 mg once daily. As needed, the dose can be slowly increased to 0.4 or a maximum of 0.6 mg. The dose must be reduced to 0.2–0.4 mg/day in patients with moderate renal failure (GFR of 30–60 mL/min) and the drug should not be used at GFR values less than 30 mL/min |
Rilmenidine – 1 mg once daily, if necessary to be increased to 2 mg in one oral dose. At GFR values <15 mL/min, it should be given in a dose of 1 mg every other day |
Overdose |
Large overdoses can paradoxically increase BP particularly with clonidine |
39.3.3 α-Methyldopa
Methyldopa was first synthesized in 1955 from amino acid derivatives of phenylalanine for treating endocrinologically active neoplastic diseases. From the early 1960s to the late 1970s, α-methyldopa remained a widely used drug in the treatment of all stages of hypertension, in part, because other therapeutic options were lacking [7]. When used in doses ranging from 250 mg to 2.0 g/day, it effectively reduces supine BP without significant orthostatic BP changes. In the more long term, intravascular volume tends to expand diminishing its effectiveness and not infrequently necessitating addition of small doses of a diuretic. With the availability of mechanistically different and inherently better tolerated antihypertensive medications, α-methyldopa has fallen out of favor beyond its use in pregnancy-induced hypertension (Chap. 61) and in patients with sympathetically driven forms of hypertension who are clonidine intolerant [18]. As regards the former, α-methyldopa lacks fetal adverse effects in utero (maintains uterine perfusion, not teratogenic) and does not reduce maternal cardiac output, and uterine and/or renal blood flow [19, 20].
Methyldopa is offered in an intravenous formulation (as the parent drug ester) making it one of the antihypertensive medications parenterally available for hypertensive emergencies. The usual intravenous dose range for α-methyldopa is 20–40 mg/kg/day in divided doses given every 6 h. The treatment of hypertensive emergencies with intravenous α-methyldopa, however, is dated with its having been replaced by more effective and easier to use compounds. In patients with renal failure, urinary excretion of free methyldopa is decreased and plasma levels of methyldopa, particularly the sulfate conjugate, are increased; hence, smaller doses may be indicated in renal failure patients, and this compound is dialyzable. The intestinal absorption of α-methyldopa, and therefore its therapeutic effect, is reduced with the co-ingestion of iron. Common side effects with α-methyldopa include somnolence and depression, which may be linked to a fall in brain biogenic amines [21, 22]. Hypersensitivity reactions, including hepatitis and Coombs-positive hemolytic anemia (due to the appearance of an antibody with specificity for red cell Rh determinants), can occur with α-methyldopa. These reactions occur in 10–20 % of patients receiving ≥1 g/day of α-methyldopa over several months [23]. α-Methyldopa can be continued in the presence of a positive direct Coombs test alone unless anemia develops in which case therapy should be quickly withdrawn. A form of hepatitis with fever, eosinophilia, and increased transaminase values can occasionally develop with α-methyldopa. This is a self-limited process that remits with drug discontinuation. α-Methyldopa may also produce a drug-induced fever with accompanying flu-like symptoms. Its use can be accompanied by an enhance release of prolactin in certain patients, thereby inducing pseudolactation. α-Methyldopa and its metabolic products can interfere with certain catecholamine assays and can interfere with the action of levodopa, bromocriptine, and monoamine oxidase inhibitors.
39.3.4 Clonidine
Clonidine hydrochloride, an imidazoline derivative, was originally developed as a nasal decongestant and vasoconstrictor. Its hypotensive and bradycardic effects were first serendipitously appreciated in 1962. Oral clonidine has an onset of action within 30 min and is particularly useful for managing hypertensive urgencies; however, it is fairly short-acting requiring that it be frequently dosed in managing hypertensive urgencies [24]. A transdermal delivery system for clonidine is available, which provides a programmed daily amount of drug for 7 days. This delivery system needs to be situated on the skin for at least 1 day to achieve steady-state plasma concentrations. Because of the time delay to reach a steady state with transdermal clonidine, oral clonidine should be continued for 1–2 days after the initial patch is first applied. Even after removal of a transdermal clonidine patch, residual drug in the skin maintains the antihypertensive drug effect for 12–24 h [25]. Transdermal clonidine is best absorbed from a chest or upper arm site [26]. Transdermal clonidine is of particular utility for the management of the labile hypertensive patient who requires multiple medications, the hospitalized patient who cannot take oral medications, and the patient with prominent early morning BP surges. At equivalent doses, transdermal clonidine is more likely than oral clonidine to cause dose-dependent salt and H2O retention [17].
Clonidine can suppress sinus and atrioventricular nodal function, which will sometimes result in significant bradycardia. Also, patients with CKD and sinus node dysfunction are at risk of developing significant bradycardia with clonidine and this drug is best avoided in such individuals [27]. If clonidine is suddenly discontinued during treatment with high doses (usually >1 mg but sometimes lower doses), rebound hypertension may occur [28]. The rebound increase in blood pressure can be quite significant in the occasional patient being treated with clonidine and is characterized by an increase in adrenergic discharge in the setting of upregulated adrenoceptors. Rebound hypertension may be more prominent if therapy with a β-blocker is ongoing when clonidine is discontinued. Such a rebound phenomenon has not been seen with moxonidine and rilmenidine [8, 9]. Clonidine overdose can result in paradoxical hypertension if the vasodepressor effect of central α2-adrenergic stimulation is exceeded by the pressor effects of peripheral α2-adrenergic receptor stimulation [29].
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


