Cellular, Molecular, and Structural Changes During Cardiac Remodeling
Richard D. Patten
Heart failure is associated with activation of neurohormonal and cytokine signaling pathways that alter the structure and function of cardiac myocytes and nonmyocyte cells. These cellular alterations culminate in the gross morphological changes in cardiac structure termed remodeling, a maladaptive process that contributes to further left ventricular dysfunction and heart failure progression. This chapter will review the cellular basis for cardiac remodeling and the mechanisms that contribute to these cellular abnormalities and, more broadly, to the pathophysiology of heart failure and its progression.
The Clinical Syndrome of Ventricular Remodeling
Left ventricular (LV) remodeling refers to alterations in ventricular mass, chamber size, and geometry that result from myocardial injury, pressure, or volume overload. The ultrastructural changes of the remodeled ventricle are the direct result of cellular alterations that include myocyte hypertrophy, myocyte apoptosis, fibroblast proliferation, and the abnormal infiltration of mononuclear inflammatory cells. This complex, progressive, and maladaptive process promotes ongoing ventricular dilatation and dysfunction contributing directly to heart failure progression.
Because ischemic heart disease is a major risk factor for the development of heart failure, much of our understanding of LV remodeling in humans has come from the study of patients following myocardial infarction (MI). In the post-MI heart, the extent of LV chamber enlargement is directly related to infarct size (1,2). In the early period (days) following a moderate to large MI, the LV cavity enlarges because of infarct expansion (i.e., elongation and thinning of the infarcted segment) (3,4,5). However, progressive LV dilatation often continues over months to years (1,2,6,7), resulting primarily from elongation or eccentric hypertrophy of noninfarcted wall segments (5,8). Figure 8-1 illustrates this concept, showing outlines of LV end-diastolic and end-systolic frames from a right anterior oblique ventriculogram obtained from a patient 1 week and 1 year following a large, anterior wall MI. While the two-dimensional length measurement of the infarcted, noncontractile segment remained stable over that time period, the elongation of contractile portions of the LV resulted in further LV dilatation.
LV remodeling following MI has been shown in several studies to portend a poor prognosis (9,10,11). White et al. reported in a series of 605 patients 1 to 2 months after MI that the strongest independent predictor of survival was LV end-systolic volume (LVESV) (12). In the angiographic substudy of the Global Utilization of Streptokinase and t-PA for Occluded Coronary Arteries (GUSTO) I Trial, LVESV of ≥ 40mL/m2was independently associated with an increase in mortality among 1,300 patients treated with thrombolytic therapy for an acute transmural MI (13). More recently, Bolognese et al. reported that among acute MI patients having undergone successful reperfusion therapy with primary percutaneous coronary intervention, 30% developed significant LV dilatation over 6 months [defined as an increase in LV end-diastolic volume (EDV) of greater than 20% above baseline] (14). As shown in Figure 8-2, patients in this study who exhibited LV remodeling had a significantly greater incidence of heart failure and death compared to patients without LV remodeling. Consistent with prior studies, 6-month LVESV was identified as a significant predictor of mortality by multivariate analysis.
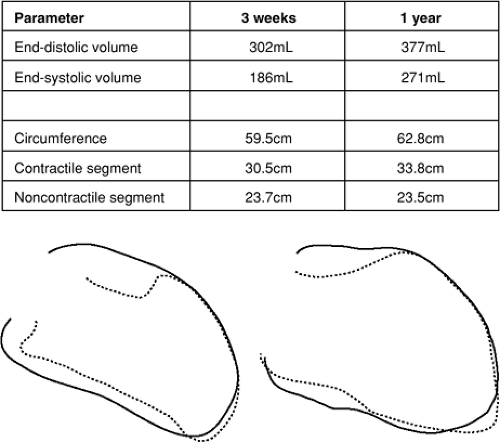 Figure 8-1 Late ventricular enlargement in a patient with anterior myocardial infarction. Marked increase in volume results from increased circumference and sphericity. The late change in circumference is due to lengthening of contractile tissue rather than further expansion of the infarcted, noncontractile segment. (Reproduced from Mitchell GF, Lamas GA, Vaughan DE, et al. Left ventricular remodeling in the year after first anterior myocardial infarction: a quantitative analysis of contractile segment lengths and ventricular shape. J Am Coll Cardiol. 19:1136–1144 , with permission.) |
Recently, two groups observed a strong association between progressive LV dilatation following MI and the incidence of sudden cardiac death and ventricular arrhythmias (15,16). These studies demonstrate that post-MI remodeling remains clinically relevant in the era of reperfusion therapy and that a direct correlation exists between remodeling and cardiac events. These data therefore support that LV remodeling is a maladaptive process leading to the gradual decline in LV function and, hence, heart failure progression.
Apart from the immediate post-MI setting, patients with chronic heart failure due to chronic ischemic or nonischemic etiologies also exhibit progressive LV chamber enlargement, which has been observed in placebo arms of LV remodeling substudies of clinical heart failure trials (17,18,19,20,21). For example, in the placebo arms of both the radionuclide and echocardiographic substudies of the Studies of Left Ventricular Dysfunction (SOLVD) trial, progressive LV dilatation and trend for increased LV mass were observed at 1 year. Thus, in addition to the post-MI setting, patients with LV systolic dysfunction with or without symptomatic heart failure exhibit progressive LV remodeling (17,18,19).
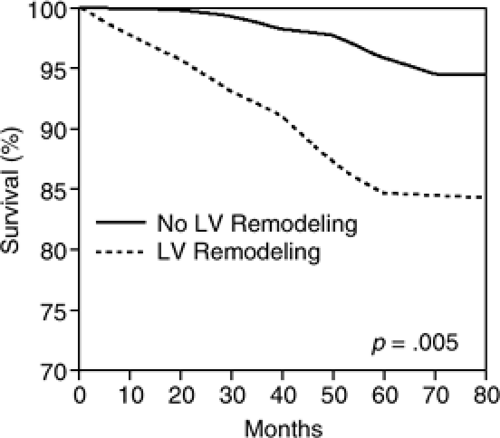 Figure 8-2 Kaplan-Meier survival curves for cardiac death in patients with and without LV remodeling at 6 months after AMI. (From Bolognese L, Neskovic AN, Parodi G, et al. Left ventricular remodeling after primary coronary angioplasty: patterns of left ventricular dilation and long-term prognostic implications. Circulation. 106:2351–2357 , with permission.) |
Cellular Alterations in Cardiac Remodeling
The clinical data previously described demonstrate that LV remodeling occurs in patients with LV systolic dysfunction and is associated with a worse prognosis. The macroscopic changes in ventricular size and shape stem from alterations in the cellular constituents of the myocardium that act in concert to bring about global LV dilatation and dysfunction and, hence, heart failure progression. This section will review the structural and biochemical alterations in cardiac myocytes and fibroblasts, and how the infiltration of inflammatory cells contributes to these pathological changes.
Cardiomyocytes
Among the cell types that exist within the myocardium, much attention (recent and remote) has focused on the cardiac myocyte. Increased myocyte size contributes importantly to the gross morphological features of ventricular enlargement. In a rat model of MI, Anversa et al. used histomorphometry to measure myocyte size distant from the infarct zone, and found that myocytes increase in length and diameter (22,23). In isolated myocytes from patients with ischemic cardiomyopathy, the Gerdes Lab (24) reported that myocyte length increased by approximately
40%, corresponding directly with the increase in LV chamber diameter. Figure 8-3 displays marked enlargement of isolated cardiomyocytes from cardiomyopathic human hearts (lower panel) compared with that obtained from controls (upper panel). Using the spontaneously hypertensive heart failure-prone rat model, the Gerdes Lab (25) showed further that increases in myocyte length during the transition to overt heart failure correlated directly with the degree of LV chamber dilatation (Fig. 8-4). Similarly, Anand et al. demonstrated in a rat MI model that myocyte length correlated directly with LV volume measured via passive pressure-volume curves obtained postmortem (26).
40%, corresponding directly with the increase in LV chamber diameter. Figure 8-3 displays marked enlargement of isolated cardiomyocytes from cardiomyopathic human hearts (lower panel) compared with that obtained from controls (upper panel). Using the spontaneously hypertensive heart failure-prone rat model, the Gerdes Lab (25) showed further that increases in myocyte length during the transition to overt heart failure correlated directly with the degree of LV chamber dilatation (Fig. 8-4). Similarly, Anand et al. demonstrated in a rat MI model that myocyte length correlated directly with LV volume measured via passive pressure-volume curves obtained postmortem (26).
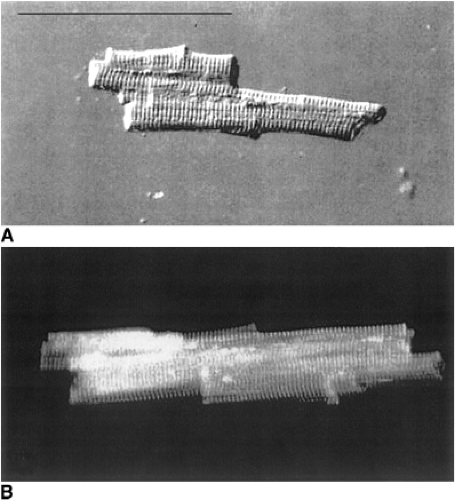 Figure 8-3 (A) Typical myocyte isolated from nonfailing human left ventricle. Bar = 100 μm. (B) Myocyte isolated from the LV of a patient with ischemic cardiomyopathy. (From Gerdes AM, Kellerman SE, Moore JA, et al. Structural remodeling of cardiac myocytes in patients with ischemic cardiomyopathy. Circulation 86:426–430 , with permission.) |
These data support that LV chamber dilatation is driven largely by myocyte enlargement, perhaps refuting the classically held notion of so-called myocyte slippage to account for LV dilatation in the failing ventricle. Myocyte slippage refers to the sliding of individual myocytes and myocyte bundles along the long axis that results from disruption of fibrillar collagen connections among these cells and bundles. The notion of myocyte slippage is based upon observations that myocyte numbers decrease across transmural segments of the LV in animal models of remodeling (27,28). Although such evidence is indirect, more recent data using scanning electron microscopy to obtain detailed structural analysis of myocytes and myocyte bundles support that myocyte slippage is likely to be a relevant phenomenon in the remodeling and dilating ventricle.
In the adult, pathological myocyte hypertrophy is characterized by the re-expression of fetal genes (i.e., genes expressed normally in ventricular myocytes during embryonic development that become quiescent soon after birth) (29). These genes include the contractile elements, β-myosin heavy chain (β-MHC), and skeletal α-actin, as well as the non-contractile protein, atrial natriuretic peptide (ANP) (29). Meggs et al. showed that increased myocyte size following experimental MI is associated with elevated expression of skeletal α-actin mRNA (30), while others have documented elevated ANP mRNA and protein within the noninfarcted myocardium (31,32). Similarly, hearts from patients with ischemic cardiomyopathy display markedly increased myocyte size (33) and elevated levels of ANP mRNA (34) in areas distant from infarction. As myocytes enlarge, they also express increasing amounts of brain natriuretic peptide (BNP) mRNA.
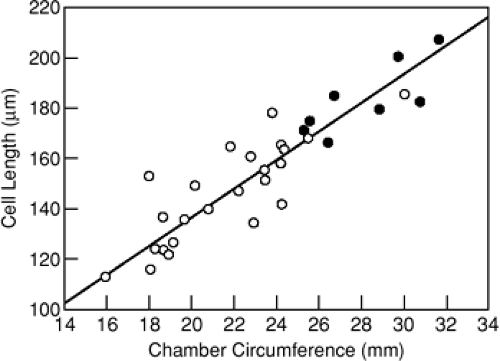 Figure 8-4 Linear regression analysis of changes in cell length and chamber circumference in aging SHHF rats. Cell length increased linearly with chamber circumference. (r = 0.93; p < 0.001; y = 5.72x + 22.1). (From Tamura T, Onodera T, Said S, et al. Correlation of myocyte lengthening to chamber dilation in the spontaneously hypertensive heart failure (SHHF) rat. J Mol Cell Cardiol. 30:2175–2181 .) |
In addition to the pathological growth of cardiomyocytes, it has become increasingly evident over the past ten years that ongoing cardiomyocyte loss contributes to progression of ventricular remodeling and dysfunction. While there exists some controversy as to which mode of cell death, necrosis, autophagy, or apoptosis predominates among cardiomyocytes in the failing heart (35), mounting evidence supports that programmed cell death (apoptosis) contributes significantly to cardiomyocyte cell loss (36). Apoptosis is a tightly regulated, adenosine triphosphate (ATP)-dependent process in which intracellular proteolytic enzymes (termed caspases) are activated, causing nuclear chromatin condensation and organized disassembly of the cell. Surrounding cells or macrophages then phagocytose resulting cell fragments so that little or no classical inflammatory response occurs (37,38,39,40,41,42).
Apoptosis Signaling Pathways
Apoptosis occurs following the activation of two general pathways: (a) The extrinsic pathway that involves agonist binding to death receptors; and (b) the intrinsic or mitochondrial pathway, which is activated by disruption of the intracellular chemical milieu caused by accumulation of reactive oxygen species, hypoxia, and/or sudden increases in intracellular Ca2+ that lead to loss of integrity of the outer mitochondrial membrane, resulting in the cytoplasmic release of pro-apoptotic initiating factors (38,39).
Extrinsic Pathway
Apoptosis can be initiated through activation of death receptors (e.g., Fas) by death-inducing ligands (e.g., Fas ligand) that lead to the recruitment of death domain-containing adaptor proteins such as Fas-associated via death domain (FADD), and the formation of a membrane-associated, death-inducing signaling complex (DISC). FADD then recruits procaspase 8 into the complex in the form of a dimer, leading to its cleavage and activation; caspase 8 then cleaves and activates procaspase 3 to its active form, caspase 3 (Fig. 8-5) (43). Caspase 8 also activates the pro-apoptotic Bcl-2 family member, Bid (see Bcl-2 Protein Family, next page), after which the C-terminal portion of Bid (tBid) translocates to the mitochondria and inserts into the outer mitochondrial membrane, facilitating the formation of the mitochondrial permeability transition pore (MPTP). This leads to the release of cytochrome C and other pro-apoptotic factors into the cytoplasm. In this manner, the extrinsic pathway is mechanistically linked to the intrinsic or mitochondrial pathway.
Intrinsic Pathway
The intrinsic pathway of programmed cell death (38) is triggered by the accumulation of reactive oxygen species, sudden increases in intracellular calcium, or hypoxic insults (44). Following these stimuli, the outer mitochondrial membrane becomes disrupted via mechanisms that are not entirely understood, though mitochondrial localization of pro-apoptotic Bcl-2 family members leading ultimately to the formation of the MPTP appears crucial in initiating this process. Upon outer mitochondrial membrane disruption, pro-apoptotic factors are released from the intermembrane space into the cytosol. The heme-containing electron transport protein, cytochrome C, is one pro-apoptotic factor released from the intermembrane space. Once in the cytoplasm, cytochrome C binds to and induces the oligomerization of apoptosis protease activating factor (Apaf-1) that recruits procaspase 9, forming a complex known as the apoptosome. Upon recruitment to the apoptosome, procaspase 9 is cleaved and activated; caspase 9 then binds and cleaves procaspase 3, resulting in caspase 3 activation. Caspase 3 is an important terminal effector of apoptosis, cleaving substrate proteins and activating other downstream caspases within the cell (Fig. 8-5).
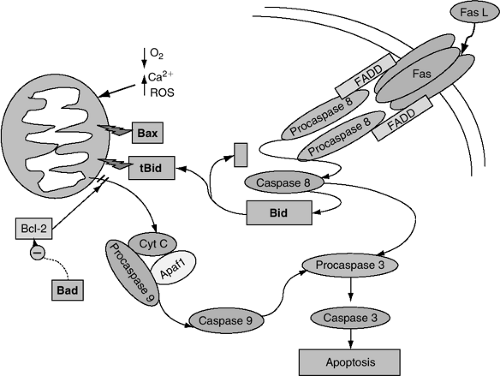 Figure 8-5 Apoptosis signaling pathways. Two major pathways converge on caspase 3, triggering apoptosis. Activation of the extrinsic pathway occurs by activation of the death receptor, Fas, by Fas lig-and (Fas L). This causes cleavage and activation of procaspase 8 to caspase 8, which cleaves the pro-apoptotic protein, Bid, that contributes to mitochondrial disruption. Caspase 8 also activates caspase 3, which then cleaves target proteins and activates other downstream caspases. The intrinsic or mitochondrial pathway is activated by stimuli such as hypoxia, reactive oxygen species, or sudden increases in intracellular calcium causing disruption of the mitochondrial membrane via activation of pro-apoptotic family members such as Bax and Bid, leading to the formation of the mitochondrial permeability transition pore (MPTP). This results in cytoplasmic leakage of intermembrane contents including the pro-apoptotic factor, cytochrome C (Cyt C), that binds and activates apoptosis protease activating factor-1 (apaf-1), causing sequential activation of caspases 9 and 3. Pro-survival Bcl-2 family members, including Bcl-2 and Bcl-xl, protect the cell from apoptosis by inhibiting mitochondrial pore formation. |
Bcl-2 Protein Family
The Bcl-2 family of apoptosis-related proteins is evolution-arily conserved, consisting of both pro-survival and pro-apoptotic factors (44,45,46). The pro-survival members of the
Bcl-2 family (Bcl-2 and Bcl-xl) inhibit apoptosis, in part, by maintaining mitochondrial membrane integrity and preventing the release of pro-apoptotic factors from the inter-membrane space (e.g., cytochrome C). The anti-apoptotic Bcl-2 proteins have been shown to inhibit the pro-apoptotic members via direct protein-protein interactions. Pro-apoptotic proteins within the Bcl-2 family, such as Bad, are composed of a Bcl-2 homology (BH3) domain that induces heterodimerization with Bcl-2 and Bcl-xl, thereby blocking their anti-apoptotic effects. Other BH3 containing Bcl-2 family members such as Bid and Bax exert their pro-apoptotic effects by inducing the formation of the MPTP. Although the precise mechanisms are not clear, activation of pro-apoptotic Bid and Bax may form a protein-permeable pore in the outer mitochondrial membrane.
Bcl-2 family (Bcl-2 and Bcl-xl) inhibit apoptosis, in part, by maintaining mitochondrial membrane integrity and preventing the release of pro-apoptotic factors from the inter-membrane space (e.g., cytochrome C). The anti-apoptotic Bcl-2 proteins have been shown to inhibit the pro-apoptotic members via direct protein-protein interactions. Pro-apoptotic proteins within the Bcl-2 family, such as Bad, are composed of a Bcl-2 homology (BH3) domain that induces heterodimerization with Bcl-2 and Bcl-xl, thereby blocking their anti-apoptotic effects. Other BH3 containing Bcl-2 family members such as Bid and Bax exert their pro-apoptotic effects by inducing the formation of the MPTP. Although the precise mechanisms are not clear, activation of pro-apoptotic Bid and Bax may form a protein-permeable pore in the outer mitochondrial membrane.
Role of Cardiomyocyte Apoptosis in LV Remodeling
The clinical and experimental literature is replete with evidence to support that apoptosis of cardiac myocytes contributes to remodeling (46). Elegant autopsy studies have shown that cardiomyocytes in normal hearts undergo cell death at very low rates (47,48,49,50), but the rate of apoptosis increases sharply in acutely ischemic hearts (51). Hearts from patients who died of an acute MI demonstrate a substantial increase in terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL)-positive cells in the infarct and peri-infarct zones (52,53). Figure 8-6 demonstrates an example of TUNEL-positive cardiomyocytes and nonmyocyte cells in the peri-infarct zone from an experimental model. Abbate et al. reported that the degree of cardiomyocyte apoptosis within the region remote from the infarct in patients who died late (>10 days) following MI correlated with greater LV remodeling. (54) These human studies provide substantial evidence that cardiomyocyte apoptosis contributes to cell death in acute myocardial injury and that the extent of apoptosis correlates with LV remodeling.
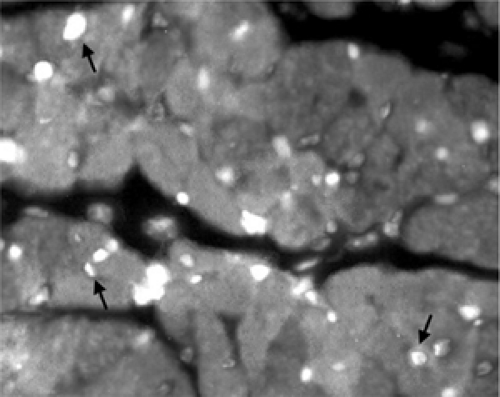 Figure 8-6 Cardiomyocyte apoptosis in the peri-infarct zone. Examples of terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) staining of the peri-infarct zone in myocardial sections from mice 24 hours following permanent coronary ligation. TUNEL-positive nuclei are bright white. Arrows indicate examples of TUNEL-positive cardiomyocyte nuclei. Numerous TUNEL-positive nonmyocyte nuclei are also seen. |
Several studies have demonstrated evidence of cardiomyocyte apoptosis in chronically failing human hearts. In cardiac biopsy samples, Saraste et al. demonstrated increased cardiomyocyte apoptosis in the myocardium of patients with dilated cardiomyopathy and heart failure. In this study, the percent of apoptotic cardiomyocytes correlated with the rapidity of heart failure progression (55). Narula et al. also reported increased TUNEL-positive cardiomyocytes in failing human myocardium and demonstrated increased levels of cytoplasmic cytochrome C and activated caspase 3 as compared to normal controls (56). These data thus support that cardiomyocyte apoptosis is present at a significantly increased rate in failing hearts and suggest that apoptotic loss of cardiac myocytes may play an important role in heart failure progression. In animal models of chronic and progressive LV dysfunction and heart failure, cardiomyocyte apoptosis occurs at an increased rate above that observed in sham or control animals. In a rat pressure overload model, Condorelli et al. demonstrated evidence of increased cardiomyocyte apoptosis following the transition from compensated hypertrophy to LV dysfunction (57). In a mouse MI model, Sam et al. noted a gradual increase in cardiomyocyte apoptosis defined both by TUNEL staining and caspase 3 activity within the noninfarcted myocardium over a span of 6 months that was associated with progressive LV dilation and heart failure (58). Both of these representative experimental studies support that progressive cardiomyocyte cell loss within the myocardium contributes to LV dilatation and progressive LV dysfunction.
Moreover, in multiple transgenic models of dilated cardiomyopathy, cardiomyocyte apoptosis is observed at an increased rate within the failing myocardium. For example, the G-protein Gq couples intracellular signaling pathways to several 7-transmembrane spanning receptors that induce cardiomyocyte hypertrophy. Overexpressing the alpha sub-unit of Gq (Gαq) in the heart leads to a significant rise in cardiomyocyte apoptosis, coupled with the development of compensated LV hypertrophy, followed by progressive LV dysfunction (45). Similarly, mice with a cardiac-targeted overexpression of tumor necrosis factor-alpha (TNF-α) develop increased cardiomyocyte apoptosis, significant LV dysfunction, heart failure, and premature death (59,60,61). These transgenic models represent a cross-section of many studies linking dilated cardiomyopathy and progressive LV dysfunction to cardiomyocyte apoptosis.
Wencker et al. (62) recently developed a transgenic mouse with cardiac-specific expression of a procaspase 8 construct that is fused to FK binding protein. This protein construct dimerizes in the presence of the pharmacologic agent, FK506. Thus, upon treatment with FK506, two procaspase 8 molecules form a dimer and their proximity leads to autocleavage and activation of caspase 8 within cardiac myocytes. These mice develop a dose-dependent increase in cardiomyocyte apoptosis, LV chamber enlargement, severe LV dysfunction, and death. One of the transgenic lines exhibited low-level caspase 8 activation that led to an apoptosis rate that was only 10-fold greater than wild type mice and was comparable to that observed in failing human hearts. These animals developed progressive LV dilation and systolic dysfunction followed by premature death, further supporting
that low-level cardiomyocyte apoptosis contributes to worsening LV function and heart failure progression.
that low-level cardiomyocyte apoptosis contributes to worsening LV function and heart failure progression.
The benefit of inhibiting apoptosis has been demonstrated in several animal models of heart failure. The broad, nonspecific caspase inhibitor IDN 1965 has been reported to prevent cardiomyocyte apoptosis and reduce cardiac remodeling in procaspase 8 transgenic mice previously described (62) and in female mice with cardiac-targeted overexpression of Gαq (63). In the rat MI model, Chandrashekhar et al. demonstrated that treatment of rats with a cell-permeable, broad caspase inhibitor Z-Asp 2,6-DCBMk immediately following coronary ligation reduced apoptosis in the remote myocardium at 4 weeks in association with a marked reduction of LV hypertrophy and dilatation (64). These data provide convincing support for the role of cardiomyocyte apoptosis in ventricular remodeling and heart failure progression.
Cardiac Fibroblasts
Ventricular remodeling is also characterized by changes within the interstitial compartment. Fibroblasts constitute approximately 90% of nonmyocyte cells in the myocardium and play a major role in the regulation of the extracellular matrix (65,66). Total interstitial collagen content is governed by the ongoing balance of synthesis and degradation (67), and myocardial fibroblasts produce types I and III collagen as well as fibronectin (68), the synthesis of which increases in response to myocardial injury (66). Increases in collagen deposition and collagen type I mRNA expression have been demonstrated within the noninfarcted myocardium of human hearts following MI (69) and in hearts from patients with chronic ischemic cardiomyopathy (33,70). Moreover, within the myocardium of patients with heart failure due to pressure overload (aortic stenosis), Hein et al. recently observed a marked increase in fibronectin; the greatest amount of fibrosis was evident in patients with heart failure and severe LV dysfunction (LVEF <30%) (Fig. 8-7) (78).
Similar findings have been observed in animal models of myocardial injury. For example, increased staining for collagen and increased collagen types I and III gene expression are well-established in the rat and mouse MI models. Fibroblasts also proliferate in response to myocardial injury. In the rat MI model, we and others observed increased nonmyocyte cell proliferation within the noninfarcted myocardium 14 days postcoronary ligation (71,72). Correspondingly, collagen content in the noninfarct zone increases and plateaus between days 7 and 14 (71), accompanied by elevations of collagen types 1 and III mRNA (73).
Upon activation, fibroblasts may also differentiate into myofibroblasts that develop phenotypic features of smooth muscle cells, expressing α-smooth muscle actin that is incorporated into contractile filaments and stress fibers. Myofibroblasts have important physiological functions in tissue injury and repair (74). Within the heart, myofibroblasts lie within the perivascular regions but, upon activation, translocate to the infarcted myocardium. In the rat MI model, myofibroblasts are evident within 3 days of injury and remain abundant through week 4, where they participate in the laying down of types I and III collagen and promote scar formation and contraction (75). Whether myofibroblasts infiltrate into regions remote from the infarct zone has not been firmly established. However, in models of cardiomyopathy induced by infusions of isoproterenol or angiotensin II (Ang II), myofibroblasts have been noted in areas of microfibrosis, presumably at sites of myocyte degeneration and loss (76). Thus, within the myocardium, contractile properties of myofibroblasts coupled with enhanced production of extracellular matrix material may contribute to increased stiffness of the left ventricle and, hence, lead to diastolic filling abnormalities.
Myofibroblasts have also been shown to stain positively for TGF-β1, angiotensin converting enzyme (ACE), and angiotensin II within the myocardium (73). Thus, myofibroblasts are also an important reservoir for components of the renin-angiotensin system and TGF-β signaling pathway.
Macrophages
As is evident from the previous discussion, alterations in myocyte and fibroblast biology in the progression of LV remodeling have been studied extensively, yet the potential role of inflammatory cells in the pathogenesis of remodeling has only recently gained recognition. Inflammatory cells play a clear role in infarct healing. After an MI, infiltration of polymorphonuclear neutrophils, macrophages, and other mononuclear cells (including mast cells) occurs rapidly within the infarct zone. However, as infarct healing is completed over several weeks, increased numbers of inflammatory cells appear within the noninfarcted myocardium. In hearts from patients with ischemic and idiopathic dilated cardiomyopathy, a fourfold increase in the number of macrophages per unit area (CD68-positive staining cells) are evident within the noninfarcted regions of the myocardium (77). Similar increases in macrophages have been observed in patients with heart failure due to aortic stenosis (78).
The infiltration of macrophages into the myocardium has also been observed in animal models of heart failure and remodeling. For example, in a rat pressure overload model, Shioi et al. (Fig. 8-8) observed a fourfold increase in interstitial macrophages following the transition to heart failure in association with greater interleukin-1β (IL-1β) and monocyte chemoattractant and activator protein 1 expression (MCAP1 or MCP-1) (79). In a rat MI model, Ono et al. (80) similarly observed increased numbers of macrophages in the noninfarct zone, many of which stained strongly for IL-1β, supporting that myocardial macrophages in this model may contribute to enhanced production of stress-activated cytokines which contribute further to abnormalities in cardiac myocytes and fibroblasts within the failing heart. The number of macrophages increased by approximately fourfold in the myocardium from Dahl salt-sensitive rats with heart failure compared to rats that did not develop heart failure. As in the rat MI model, the interstitial macrophages in this model stained positively for IL-1β, further emphasizing that macrophages are likely a significant if not predominant source of cytokines in these models (79).
Hayashidani et al. recently examined the remodeling effects of gene therapy directed against MCP-1, inhibited by a dominant negative construct expressed using replication-deficient, adenoviral-mediated gene transfer into the limb muscles of these mice. Following MI, wild type mice
exhibited potent induction of MCP-1 gene expression within the infarct and noninfarct zones that waned over the ensuing 4 weeks. Mice expressing the dominant negative MCP-1 developed less infiltration of macrophages within the noninfarct zone associated with diminished expression of both TNF-α and TGF-β1, coinciding with reduced fibrosis and myocyte hypertrophy (Fig. 8-9) (81). These findings therefore support that inhibiting mononuclear cell infiltration within the noninfarct zone limits cytokine gene expression and cardiac remodeling.
exhibited potent induction of MCP-1 gene expression within the infarct and noninfarct zones that waned over the ensuing 4 weeks. Mice expressing the dominant negative MCP-1 developed less infiltration of macrophages within the noninfarct zone associated with diminished expression of both TNF-α and TGF-β1, coinciding with reduced fibrosis and myocyte hypertrophy (Fig. 8-9) (81). These findings therefore support that inhibiting mononuclear cell infiltration within the noninfarct zone limits cytokine gene expression and cardiac remodeling.
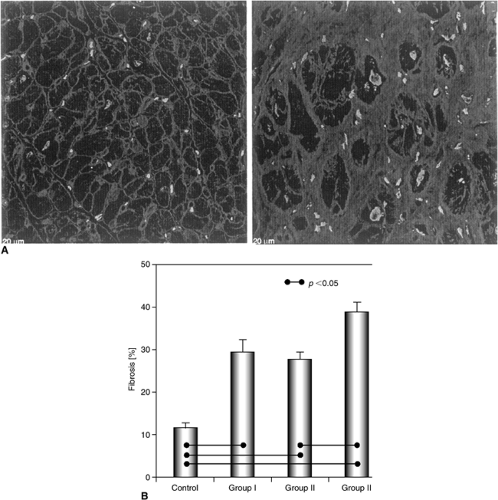 Figure 8-7 Marked fibrosis in sections from hypertrophied human hearts due to aortic stenosis. Intercellular fibronectin framework is selectively stained (nuclei are slightly brighter). (A) Left: Normal myocardium shows fine septa between unstained myocytes; Right: severe fibrosis with few myocytes. (B) Bar graphs representing area percent of myocardial fibrosis determined by fibronectin staining. Group I (preserved LVEF) and group II (LVEF ≥30% <50%) demonstrate increased fibronectin staining. Most severe fibrosis is seen in group 3 (LVEFs <30%). (From Hein S, Arnon E, Kostin S, et al. Progression from compensated hypertrophy to failure in the pressure-overloaded human heart: structural deterioration and compensatory mechanisms. Circulation. 107:984–991, with permission.) |
Mast Cells
Similar to macrophages, the role of mast cells in LV remodeling is not completely understood but these mononuclear inflammatory cells have recently been shown to contribute
to extracellular matrix and myocyte remodeling in the failing heart. Patella et al. (82) demonstrated in samples from human dilated cardiomyopathic hearts a fourfold increase in mast cell density compared with normal control samples. Moreover, Petrovic et al. observed a twofold increase in mast cell density in human dilated cardiomyopathy, while a greater number of mast cells was observed in myocarditis (83). These studies in human tissue support that mast cells are recruited to the myocardium in response to chronic myocardial injury.
to extracellular matrix and myocyte remodeling in the failing heart. Patella et al. (82) demonstrated in samples from human dilated cardiomyopathic hearts a fourfold increase in mast cell density compared with normal control samples. Moreover, Petrovic et al. observed a twofold increase in mast cell density in human dilated cardiomyopathy, while a greater number of mast cells was observed in myocarditis (83). These studies in human tissue support that mast cells are recruited to the myocardium in response to chronic myocardial injury.
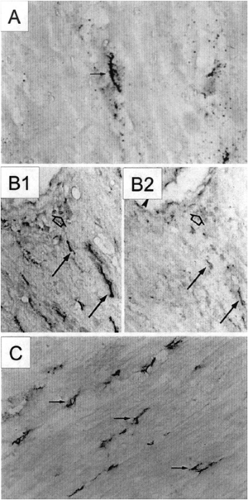 Figure 8-8 (A) In 11-week-old DS rats with heart failure, interstitial cells (arrow) stained positive for IL-1β. Original magnification 800x. (B) Serial sections of heart tissue stained with antibodies to macrophages (B1) or to IL-1β (B2) are shown. The interstitial cells (arrows) and perivascular cells (open arrows) positive for IL-1β were predominantly macrophages. Original magnification 220x. (C) The number of macrophages was increased in the hearts of DS rats with heart failure. Original magnification 200X. (From Shioi T, Matsumori A, Kihara Y, et al. Increased expression of interleukin-1β and monocyte chemotactic and activating factor/monocyte chemoattractant protein-1 in the hypertrophied and failing heart with pressure overload. Circ Res. 81:664–671, with permission.)
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|