Cellular and Molecular Abnormalities in Failing Cardiac Myocytes
Rachel Wilson
Xiongwen Chen
Steven R. Houser
Congestive heart failure (CHF) is a syndrome characterized by poor pump performance of the heart, which initially leads to decreased exercise tolerance but eventually progresses to involve pulmonary and systemic congestion. It is the ultimate outcome of many cardiovascular diseases, including hypertension, ischemic coronary disease, idiopathic myopathies, and valvular disorders. Remodeling of the failing heart at the organ, cellular, and molecular levels leads to progressive deterioration of cardiac pump function and arrhythmia, the major causes of morbidity and mortality in heart failure (HF) patients. In this chapter, we will focus on changes that occur in individual cardiac myocytes at the cellular and molecular levels that cause depressed cardiac contractility (the ability of the muscle to develop force and shorten) and lethal arrhythmias. The knowledge on this topic is substantial and has been obtained from studies of failing human myocytes and tissues and from failing myocytes from animal models in which heart failure was induced by increasing hemodynamic stress (pressure and volume overload), occlusion of coronary arteries (myocardial infarction), rapid pacing, or transgenetic approaches (gene overexpression or knockout) in mice. Throughout the chapter we will identify areas of consensus and those that still are unresolved. In addition, we will briefly discuss the most recent advances on the idea that CHF can result from excess myocyte death and cardiac repair may be possible by generating new myocardium.
Decreased Myocyte Contractility Reserve is Centrally Involved in Heart Failure
Depressed contractility of cardiac myocytes plays a critical role in heart failure. In the mildest early stages of HF, slowing of myocyte contraction and relaxation rates and prolongation of the action potential duration have been consistently observed (1). In advanced HF, force production and shortening magnitude are normal or even greater than normal (2) at slow beating rates (<30 per minute), but decrease (or remain constant) when heart rate increases (negative force-frequency relationship). This is contrary to the relationship in nonfailing myocytes, where increasing the heart rate causes force production and shortening magnitude to increase (positive force-frequency relationship) (2). The responses of failing myocytes to high extracellular Ca2+ and adrenergic stimulation are also significantly reduced. These studies show that the fundamental cellular alterations of failing myocytes allow for contractile function to be maintained at rest, when the heart rate is slow, but lead to the inability of the myocytes to increase their contractile performance (contractility) when demand for additional cardiac output is increased. We and others have termed this phenomenon depressed contractility reserve.
Abnormal Ca2+ Handling is Primarily Responsible for Decreased Myocyte Contractility
With each heartbeat, depolarization of the myocyte surface membrane causes opening of Ca2+ channels that reside in the transverse tubules. The subsequent Ca2+ influx induces Ca2+ release from the sarcoplasmic reticulum (SR). The resultant increase in cytosolic [Ca2+] (the so-called Ca2+ transient) activates myofibrillar proteins to cause contraction. The force and speed of contraction are regulated by varying these Ca2+-dependent processes.
Myocyte contractility is depressed in CHF, with the fundamental defect being an inability to increase force much beyond that exhibited at resting levels. By studying animal models and explained failing human hearts, multiple causes of depressed myocyte contractility in heart failure have been revealed, including altered
cytoskeleton (3); changes in myofilament function human CHF (4); animal CHF (5); altered β-adrenergic signaling (6); and abnormal myocyte Ca2+ regulation and excitation-contraction coupling (ECC) (1). In this chapter, we will focus on alterations in myocyte electrophysiological and contractile function in CHF, with a concentration on those factors producing alterations in Ca2+ regulation.
cytoskeleton (3); changes in myofilament function human CHF (4); animal CHF (5); altered β-adrenergic signaling (6); and abnormal myocyte Ca2+ regulation and excitation-contraction coupling (ECC) (1). In this chapter, we will focus on alterations in myocyte electrophysiological and contractile function in CHF, with a concentration on those factors producing alterations in Ca2+ regulation.
One of the common observations made in failing cardiac myocytes from both human patients and animal models is that the action potential (AP) is prolonged (Fig. 2-1). There are multiple contributors to AP prolongation, with reduced repolarizing potassium (K+) currents playing a central role, as discussed later. Parallel to the prolonged APs, the Ca2+ transient is also prolonged in myocytes from explanted hearts of human HF patients (7) and animal CHF models. At very slow pacing frequencies, the relatively normal contractility seen in cardiac myocytes from human patients with end-stage cardiac disease corresponds with a maintained amplitude of the Ca2+ transient similar to that seen in nonfailing human myocytes. However, the amplitude of the Ca2+ transient is decreased [in human (7) and animal models (8)] at physiological pacing rates. In addition, diastolic Ca2+ concentration becomes elevated (9) and the decay of the Ca2+ transient is slowed (7,9). These changes contribute to diastolic dysfunction and eventually to poor systolic (pump) performance.
The regulation of the Ca2+ transient is abnormal in failing cardiac myocytes. In normal cardiomyocytes, increasing the beating rate causes an increase in the amplitude of the Ca2+ transient and the force of contraction (positive force-frequency relationship) (Fig. 2-2). In myocytes with mild to moderate hypertrophy, the peak systolic Ca2+ is normal under basal conditions (e.g., low stimulation frequency), but becomes depressed when conditions that increase cellular Ca2+ loading are imposed (faster pacing rates, high extracellular Ca2+ or catecholamine exposure). In severely failing myocytes (e.g., myocytes from end-stage human heart failure), peak systolic Ca2+ and force (or shortening) are both close to normal at very slow pacing rates. However, when the beating rate is increased there is
either no change or a decrease in peak systolic Ca2+ and force of contraction (negative force-frequency relationship). Furthermore, diastolic Ca2+ in failing myocytes increases more than in nonfailing myocytes. These results strongly support the idea that changes in myocyte Ca2+ handling are a final common pathway for progressive deterioration of cardiac pump function in CHF. More support for this notion comes from studies that show that the transition from compensated hypertrophy to the early stages of CHF occurs when myocytes first lose their ability to normally maintain Ca2+ balance, and correction of Ca2+ handling defects is able to delay or prevent this transition (10).
either no change or a decrease in peak systolic Ca2+ and force of contraction (negative force-frequency relationship). Furthermore, diastolic Ca2+ in failing myocytes increases more than in nonfailing myocytes. These results strongly support the idea that changes in myocyte Ca2+ handling are a final common pathway for progressive deterioration of cardiac pump function in CHF. More support for this notion comes from studies that show that the transition from compensated hypertrophy to the early stages of CHF occurs when myocytes first lose their ability to normally maintain Ca2+ balance, and correction of Ca2+ handling defects is able to delay or prevent this transition (10).
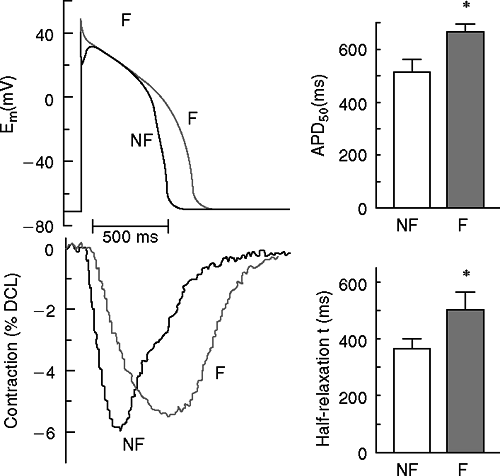 Figure 2-1 Action potential and contraction changes in heart failure. Representative action potentials (top) and contraction (bottom), measured as % of diastolic cell length from nonfailing (NF) and failing (F) myocytes. Action potential duration and contraction duration were significantly longer in failing (F) than nonfailing (NF) myocytes. All myocytes were paced at 0.5 Hz. |
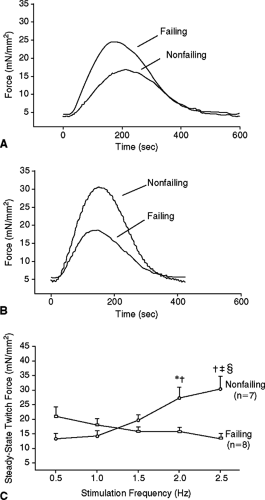 Figure 2-2 Force-frequency relationship in nonfailing and failing myocardium. Steady-state twitch tracings (1.75 mM Ca2+) of trabeculae from failing and nonfailing human hearts at (A) 0-5 Hz and (B) 2.5 Hz are represented. (C) Average steady-state twitch force as a function of stimulation frequency in failing and nonfailing myocardium. The positive relationship in nonfailing human ventricular muscle is reversed in failing muscle. * p <0.05 when compared to within-group twitch force at 1.0 Hz; † p <0.01 when compared to within-group twitch force at 0.5 Hz; ‡ p <0.01 when compared to within-group twitch force at 1.0 Hz; § p <0.05 when comparing between-groups twitch force. |
Abnormal Excitation-Contraction Coupling in Failing Myocytes
Ca2+ transients (dynamic cytosolic Ca2+ changes) are brought about by a combination of Ca2+ influx and SR Ca2+ release. When a cardiac myocyte is depolarized, an action potential is elicited and Ca2+ enters the myocyte down its transmembrane electrochemical gradient through the L-type calcium channel (LTCC). This Ca2+ influx, termed the Ca2+ current, ICa, then triggers a larger amount of Ca2+ to be released from the SR by activating the Ca2+ release channel (ryanodine receptor, RyR) through a mechanism termed calcium-induced calcium release (CICR) (11,12). The efficacy of ICa as a trigger to induce SR Ca2+ release is termed the ECC gain. These processes cause the free cytosolic Ca2+ concentration to increase rapidly and Ca2+ binds to the thin filament protein troponin C to induce contraction. The rate and magnitude of myocyte contraction (force and shortening) are largely determined by the amount of Ca2+ entering the cytoplasm with each systole. The duration of contraction and the rate of relaxation are primarily determined by processes that reduce cytosolic [Ca2+]. Cytosolic Ca2+ is lowered by three principal processes: sequestration back into the SR via SR Ca2+-ATPase (SERCA), transport out of the cell via the sarcolemmal Ca2+-ATPase, and transport out of the cell by the Na+/Ca2+ exchanger (NCX) (Fig. 2-3, ECC). In a steady state, the Ca2+ that enters with each action potential is removed from the cell primarily by the NCX, and the Ca2+ that is released from the SR is resequestered there via SERCA. The amplitude and duration of the Ca2+ transient are regulated in the normal heart to orchestrate changes in myocyte contractility. In CHF this regulation is abnormal and results in Ca2+ transients of reduced size and prolonged duration. Decreased SR Ca2+ release is now known to be the principal reason for the reduced size of the Ca2+ transient in HF. Significantly, it appears that multiple cellular defects can contribute to the reduced SR Ca2+ release of the failing myocyte.
The ECC process is abnormal in failing myocytes from human and most animal models and contributes to the reduced size of the Ca2+ transient. In a rat hypertrophy/HF model, it has been shown that ICa-1, is a less effective trigger of SR Ca2+ release in hypertrophied and failing myocytes. This defective signaling could be rescued in hypertrophied (but not failing) myocytes by exposure to β-adrenergic agonists, which increase ICa and SR Ca2+ load. We and others have also shown that Ca2+ release from the SR in failing cardiomyocytes is disorganized and this contributes to reduced Ca2+ transient size (13,14). Furthermore, in some animal CHF models where there is no change of SR Ca2+ load, the Ca2+ transients are smaller than in normal cardiac myocytes (8). Cannell et al. (15) and Gomez et al. (8) found that in spontaneously hypertensive rats (SHRs) the ability of ICa-L to trigger calcium release from the SR in both hypertrophied and failing hearts was reduced (a reduction in ECC gain). Several models were proposed to explain these ECC defects. These include a decreased density of T-tubules, an increased distance between the sarcolemmal LTCC and RyR on the face of the junctional SR membrane, and mismatch of the numbers or properties of the LTCC and RyR. He et al. (16) reported a decrease in T-tubule density in tachycardia-induced failing canine myocytes. However, this is not supported by a recent, preliminary report in failing human myocytes. In addition, reduced ECC gain does not adequately explain the fact that Ca2+ transients are similar in normal and failing human myocytes at slow pacing rates and only become significantly different when the pacing rate increases. In this regard, a frequency-dependent abnormality in ECC, decrease in the size of ICa-L, or its efficacy as a trigger of SR Ca release could be responsible for the reduction in SR Ca2+ release at fast heart rates in failing human myocytes. The respective roles of the SR Ca2+ load and the L-type calcium current in abnormal ECC in failing myocytes will be discussed in some detail below.
β-Adrenergic Signaling
Physiologically, ECC is primarily regulated via the sympathetic neurohormones norepinephrine and epinephrine, which increase cardiac myocyte force-generating capacity and heart rate. Catecholamines mediate their chronotropic and inotropic effects by binding to the seven transmembrane spanning β-adrenergic receptors (β-ARS) (primarily β1 and β2 subtypes), which couple to adenylyl cyclase via stimulatory G-proteins to produce cAMP (Fig. 2-3). There is some evidence that β2-AR can couple to inhibitory G-proteins, but this topic will not be discussed in this chapter. Increased intracellular cAMP activates protein kinase A (PKA), which phosphorylates multiple proteins to induce increased myocyte force-generating capacity, while decreasing the duration of contraction. PKA phosphorylation of the LTCC causes an increased channel open probability and an increased Ca2+ influx (ICa-L) with each action potential. This Ca2+ increases the efficacy of EC coupling and supplies the Ca2+ needed to increase SR Ca2+ stores. Phosphorylation of phospholamban (PLB, an inhibitor of SERCA) removes its inhibition of SERCA, thereby increasing SERCA activity and causing more Ca2+ to be transported into the SR, again causing the SR Ca2+ load to increase. PKA also phosphorylates the SR Ca2+ release channel (RyR), causing an increase in RyR open probability and contributing to increased SR Ca2+ release. PKA also phosphorylates troponin C and the pacemaker-related so-called funny channel to modulate myofilament Ca2+ binding affinity and the rate of firing of pacemaker cells,
respectively. The net effect of sympathetic stimulation is to enhance myocyte contractility and relaxation by increasing the size of the Ca2+ transient and hastening the rate of its decay.
respectively. The net effect of sympathetic stimulation is to enhance myocyte contractility and relaxation by increasing the size of the Ca2+ transient and hastening the rate of its decay.
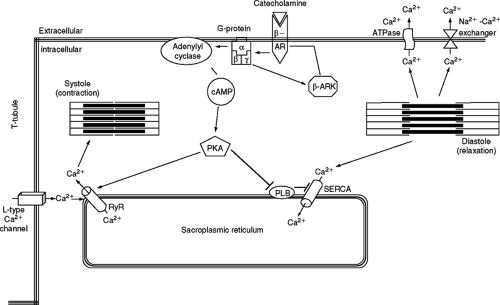 Figure 2-3 Excitation-contraction coupling and adrenergic pathway. Cardiac contraction is initiated by the influx of extracellular calcium through L-type calcium channels (LTCCs) in response to electric stimulation. The rise in intracellular calcium triggers calcium release from the sarcoplasmic reticulum (SR) via the ryanodine receptor (RyR). Calcium then associates with troponin C in the sar-comere and stimulates contraction (systole). Calcium is released from the sarcomere (diastole) and removed the cytoplasm by resequestration into the SR by the SR calcium ATPase (SERCA), extrusion from the cell by the sarcolemmal sodium-calcium exchanger (NCX), or extrusion from the cell by the sarcolemmal calcium ATPase. Activation of the β-adrenergic receptor (β-AR) by cate-cholamine binding activates adenylyl cyclase through a G-protein, generating cAMP, which activates protein kinase A (PKA). PKA phosphorylates the RyR and phospholamban (PLB), which modulates the activity of SERCA. Activation of β-AR kinase (β-ARK) by β-AR signaling creates a negative feedback loop. |
In conditions of sustained pathological hemodynamic stress that lead to HF, the sympathetic nervous system is chronically upregulated in an attempt to maintain cardiac output. This persistent sympathetic activation produces abnormalities in adrenergic signaling that reduce contractility reserve in failing cardiac myocytes. β-adrenergic signaling pathways can activate β-adrenergic receptor kinases (β-ARKs, members of the G-protein coupled receptor kinase family, GRKs), which act on the β-AR to decrease its activity in a negative feedback mechanism (17) (Fig. 2-3). Sustained exposure to β-agonists results in attenuation of signaling through this pathway by a combination of receptor phosphorylation, receptor internalization, reduced receptor protein synthesis, and increased receptor degradation (18). Phosphorylation of β-ARs by PKA or GRKs (19,20,21) induces endocytosis of agonist-bound receptors (22). For GPCRs, this can occur via two pathways: clathrin-coated pits or caveolae. β2-adrenergic receptors are internalized preferentially by the clathrin-coated pit pathway (23), though there is evidence that localization to caveolae is necessary for the physiological activity of the β2-AR (24).
β-Adrenergic Signaling in Hypertrophy and Heart Failure
A variety of disruptions in the β-adrenergic signaling pathways have been observed in heart failure. Chronic upregulation of sympathetic nervous system activity in CHF causes increased circulating catecholamines (25,26). This leads to one of the prominent features of the failing heart—desensitization and downregulation of β-ARs
(27,28). Many studies have shown that β1-AR mRNA and receptor number decrease by approximately 50% in human heart failure (29), but no change in either parameter for β2-ARs occurs (29).
(27,28). Many studies have shown that β1-AR mRNA and receptor number decrease by approximately 50% in human heart failure (29), but no change in either parameter for β2-ARs occurs (29).
Studies employing transgenic mouse models have further emphasized the important role in β1-ARs in heart failure. Overexpression of β1-ARs in mouse myocardium causes hypertrophy and interstitial fibrosis, leading to cardiac dysfunction with aging (30,31). Functional studies of these mice indicated that contractility and rate of isovolumic relaxation are initially increased in young animals. However, both systolic and diastolic function were impaired and contractility and relaxation declined steadily with age (32). It is clear that persistent overstimulation of the β1-AR in healthy mouse hearts is maladaptive, and evidence from some studies indicates that the mechanism involves apoptosis (33,34).
In contrast to the significant downregulation of β1-ARs in heart failure, there is no major change in the number of β2-ARs, though their activity is depressed due to uncoupling from adenylyl cyclase (27). The change in the β1/β2 AR ratio may point to an increased role of β2-AR activity in conditions where β1-ARs were suppressed. This is supported by studies in which β2-ARs were overexpressed. At lower levels (60- to 100-fold increase above endogenous levels), cardiac function was enhanced independent of ligand binding: contraction amplitude increased, calcium transients were greater, and relaxation times were faster in the transgenic mice than their nontransgenic littermates (35,36). However, in transgenic mice with higher myocardial β2-AR transgenic overexpression (200- to 350-fold increase above endogenous levels), there was an age-dependent progression to cardiac myopathy (35,37).
Because of the dose-dependent disparities in β2-AR, several studies have examined whether β2-AR overexpression is advantageous or deleterious in cardiac hypertrophy and failure. β2-AR-overexpressing mice subjected to transverse aortic constriction (TAC) to induce chronic pressure overload exhibited more severe heart failure than did sham-operated control animals (38), and also showed a decrease in transgene expression and in β2-AR-ligand binding (39). However, cardiac-specific β2-AR overexpression was beneficial in transgenic mice subjected to coronary ligation (myocardial infarction, MI). Infarct size and hypertrophy were comparable in transgenic and wild-type mice, but the β2-AR-overexpressing animals exhibited higher contractility, lower left ventricular end diastolic pressure (LVEDP), and a lower incidence of pleural effusion after 9 weeks of infarction (40). Our view of these data is that β2-AR signaling is cardioprotective, at least at normal or modestly increased levels of activity. The data suggesting that persistent β1 activity is ultimately harmful while β2 signaling is protective imply that these adrenergic receptors are coupled to different downstream signaling pathways. There is some evidence suggesting that β2 activity is antiapoptotic and this could be related to its beneficial role in cardiac stress states (41).
The desensitization of β-ARs in heart failure is due in part to the actions of β-AR kinase 1 (βARK1, also known as GRK2) (20,21,22,42). Cytosolic βARK binds to the Gβγ sub-unit freed by β-AR activation of the G-protein, which allows it to translocate to the plasma membrane and phosphorylate the agonist-bound β-AR (42). In the heart, βARK forms a stable cytosolic complex with phosphoinositide 3-kinase (PI3K). Translocation of βARK to the plasma membrane has been shown to facilitate PI3K movement to the membrane as well, which appears to play an important role in the internalization of the β-AR in response to cate-cholamine stimulation (43,44).
Many studies have supported the role of PI3K in cardiac hypertrophy and failure (45). Cardiac-specific constitutive activation of the p110α subunit of PI3K caused a significant increase in heart size, and constitutive activation of a downstream effector of PI3K, Akt, also increased cardiac hypertrophy (45). In response to short-term transverse aortic constriction (TAC) in the mouse, the subunit p110γPI3K (PI3Kγ) was activated without an increase in protein levels, and this activation was shown to be βARK-dependent (46,47). Cardiac-specific overexpression of an inactive PI3Kγ mutant prevented β-AR desensitization and internalization, but there is evidence that other isoforms can compensate for the loss of PI3Kγ (48).
Another characteristic of the β-adrenergic pathway in heart failure is that persistent elevation in circulating catecholamines causes a disruption in the binding of β-ARs to their respective G-proteins (27). In normal myocardium, β2-AR/Gi coupling appears to act as a brake on the β-AR/Gs/ adenylyl cyclase pathway (49,50,51,52,53). Gαi levels are increased in failing hearts (50), which, combined with the altered ratio of β1/β2-ARs as discussed earlier, indicates a prominent role in β2-AR/Gαi signaling in heart failure. This has been further supported by studies crossing transgenic mice overexpressing β2-AR with mice overexpressing an inactivated Gα i subunit. The β2-AR/heterozygous Gα i2 knockout mice developed earlier and more robust cardiac hypertrophy and earlier heart failure, and the β2-AR/homozygous Gα i2 knockout mice died within 4 days of birth (51). These data indicate an essential role for Gα i2 in protection from chronically increased β2-AR signaling (53).
There is now ample evidence that β2-AR can couple promiscuously to multiple signaling pathways (including Gα i and Gα s) and is involved in the regulation of cardiac contractility, hypertrophy, and heart failure. β2-AR activation has been shown to activate a Gi-Gβγ-PI3K-mediated survival pathway (41). The factors that regulate how, why, and when β2-ARs switch their coupling from Gs to Gi in myocytes are not well known at present but could involve signaling involving L-type Ca2+ channels and MAP kinase pathways (54,55).
β-Blockers as Therapy for Heart Failure
As previously elucidated, multiple steps in the β-adrenergic signaling pathway are disrupted in the hypertrophied and failing heart, primarily involving the downregulation and desensitization of the β-ARs. Paradoxically, however, inhibitors of the β-ARs (β-blockers), which might exacerbate the disruption of β-adrenergic signaling in heart failure and are known to precipitate negative inotropy at sufficiently high concentrations, have demonstrated a noteworthy efficiency in preventing pathological remodeling in heart failure, improving cardiac function, and even improving survival in heart failure patients (6,56). Though the mechanisms for the improvement seen with β-blockers
have not yet been completely elucidated, recent studies have provided insight into possible mechanisms (57,58).
have not yet been completely elucidated, recent studies have provided insight into possible mechanisms (57,58).
One of many possible mechanisms for improving cardiac function in CHF is the reduction of persistently activated adenylyl cyclase, thereby decreasing cAMP levels and PKA activity and correcting the phosphorylation defects of Ca2+ regulatory proteins. One of the downstream effectors of PKA is the RyR, and hyperphosphorylation of the RyR is thought to be involved in dysregulated Ca2+ in heart failure (58). Reducing the hyperphosphorylation of the RyR restores RyR function toward normal, which may partially explain the beneficial effects of β-blockers on contractility and adrenergic responsiveness in CHF patients (59). β-blockers may also help restore the energy balance in failing hearts, which are typically energy-deficient and starved for high-energy phosphates (59). Also, β-blockers have been shown to reverse some HF-specific alterations in cardiac gene expression, another possible mechanism of the beneficial effects of β-blockers (59). While the mechanisms for the beneficial effects of β-blockers are still not clear, this represents a major area for exploration for novel CHF therapies.
The L-Type Ca2+ Current (ICa-L) in Failing Myocytes
The LTCC plays multiple critical roles in the function of cardiac myocytes. The LTCC is responsible for the upstroke of the action potential (AP) in the sinoatrial node (SA) cells and contributes to the pacemaker activity of these cells. The current through the LTCC is also responsible for the conduction of the AP through the AV node and the plateau phase of the AP in the atrial, ventricular, and His-Purkinje systems (60). The major function of the LTCC in cardiac myocytes is to control the influx of Ca2+ ions that serve as the trigger for SR Ca2+ release (ECC). Ca2+ influx through the LTCC elevates the [Ca2+] in a diffusion-limited subsarcolemmal space between the T-tubules and junctional SR, which are in close proximity. Accumulation of subsarcolemmal Ca2+ promotes Ca2+ binding to and opening of the Ca2+ release channel (RyR) in the junctional SR. The Ca2+ influx through LTCC must be balanced by Ca2+ efflux through the NCX and sarcolemmal Ca2+-ATPase. Unbalanced fluxes either load or unload the SR until flux balance is restored in a new steady state.
Changes in the abundance, isoform type, regulation, or localization of the LTCC could cause abnormal Ca2+ handling in CHF. These topics have been well-studied, but the results are far from conclusive. In general, the consensus of these studies is that in mild to moderate hypertrophy, LTCC abundance and Ca2+ influx are either increased or unchanged (61,62,63). These changes are associated with a transient increase in myocyte contractility and the initiation of pathological cardiac hypertrophy. In severe left ventricular hyperthrophy (LVH) and CHF, available evidence suggests that LTCC abundance and Ca2+ influx are either unchanged (64,65,66) or reduced (67). These changes are associated with a progressive reduction in cardiac contractility and dilation of the ventricular wall. The data from animal models fall into this generalized scheme very well, but the data from terminal human heart failure patients are less uniform, possibly due to a larger variation in disease etiology and to the fact that some human studies have been performed using very poor-quality isolated myocyte preparations. Supporting our hypothesis are studies showing that dihydropyridine (DHP) binding is unchanged in idiopathic dilated hearts but is decreased in ischemic cardiomyopathy. A study performed by Takahashi et al. (67) examined DHP binding sites in CHF myocardium with four etiologies (ischemic, idiopathic, hypertensive, and congenital) and found that LTCC expression was down by ∼47% at both mRNA and DHP binding (protein) levels in all groups. These results are consistent with a study in which ICa was measured in nonfailing and failing human myocytes from our group, in which we suggest a significant reduction in LTCC density in end-stage human heart failure (68).
One study in human myocardium has explored the idea that LTCC isoform shifts occur in CHF. This study revealed the switching of splicing isoforms at segment 3 in domain IV in failing human hearts using competitive RT-PCR (69). The functional significance of these isoform shifts is largely unknown and deserve further study.
In our view, the most reliable approach to determine the density of functional LTCCs is with biophysical techniques. LTCC abundance and function have been studied at the whole-cell and single-channel levels (61,62,63). These studies suggest that in mild to moderate hypertrophy, ICa-L increases or remains constant and ICa-L kinetics do not change, whereas in severe cardiac hypertrophy and heart failure ICa-L density decreases but there are few, if any, changes of channel properties (61,62,63). In this regard, a few studies have found that ICa-L decay is slowed in HF and have suggested that this slower decay might result in larger Ca2+ influx and contribute to the prolongation of action potentials in failing cardiac myocytes (70). In a pacing-induced dog model of CHF, ICa-L density was not changed but the inactivation at positive potentials was slowed, which resulted in a higher plateau of APs (71). These ideas need further study.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


