Fig. 26.1
Onset of polymorphic VT symptomatic for syncope, converted to sinus rhythm after cardiopulmonary resuscitation
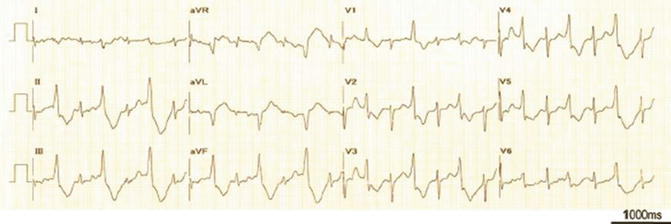
Fig. 26.2
Restoration of sinus rhythm with frequent monomorphic PVCs
Clinical Course and Therapeutic Management
This event was similar to his typical episodes. During his hospital stay, he was treated with a beta-blocker (propranolol 3 mg/kg/die) with no further episodes.
Genetic testing revealed a mutation in the cardiac ryanodine receptor gene (RyR2), consistent with CPVT.
26.3 Catecholaminergic Polymorphic Ventricular Tachycardia (CPVT)
Epidemiology
The prevalence of CPVT is about 1:10,000. The mean age of onset is between 7 and 9 years, although later onset has been reported. Approximately 30 % of patients have a family history of stress-related syncope, seizure, or SCD before age 40 years. There is a high level of penetrance of the disease (75–80 %) [1].
Clinical Presentation
Syncope triggered by exercise or emotion is the typical clinical presentation of CPVT patients and sometimes a reproducible, stress-related, bidirectional VT in the absence of a structural heart disease or QT interval prolongation could be documented. CPVT appears as one of the most malignant forms of ventricular arrhythmia. By age 40, almost 80 % of untreated CPVT patients develop symptoms (syncope, VT, or VF), and overall mortality is 30–50 % [2]. The first manifestation of the disease can be SCD in a significant number of cases [3].
Electrocardiographic Features
Brugada-like ST elevation, QT interval abnormalities, and atrioventricular and intraventricular conduction defects are absent in resting ECG of patients with CPVT. Ventricular arrhythmia distinguishing CPVT is characterized by alternating QRS axis with 180° rotation on a beat-to-beat basis (“bidirectional VT”). However, this typical arrhythmia is not observed in all patients. With increasing exercise workload, a progressive worsening of arrhythmias is a characteristic feature of CPVT. A heart rate of 110–130 beats/min is the cutoff to the appearance of ventricular arrhythmias during exercise stress testing, initially as polymorphic PVCs. Then, the complexity and frequency of arrhythmias progressively worsen as workload increases: ventricular bigeminy, run of polymorphic PVCs, and polymorphic or bidirectional VT. Bidirectional VT may degenerate into polymorphic VT and VF, if exercise is not quickly stopped. On the recovery phase, VT rate and VT duration progressively diminish until arrhythmias disappear. During exercise, at a range of heart rates similar to or slower than that of ventricular arrhythmias, isolated premature atrial complexes, nonsustained supraventricular tachycardia, and short runs of AF usually are observed [3, 4].
Diagnosis of CPVT
Resting ECG, echocardiography, and EP testing frequently are completely normal. CPVT diagnosis is clinically based on symptoms (syncope or aborted SCD), family history, and response to exercise or isoproterenol infusion. Holter monitoring, exercise, and drug provocation demonstrated ventricular arrhythmias in more than 80 % of patients. Not infrequently, syncopal episodes are considered as vasovagal in origin, and no further workup is performed. If the loss of consciousness is associated with convulsions, as it happened in our case, it may be misdiagnosed as epileptic seizures if a prolonged circulatory arrest resulted in brain ischemia [1].
When a syncopal episode induced by exercise or emotion occurs in a child or in a young patient with a normal resting ECG and no structural heart disease, CPVT should be hypothesized [1, 3].
The most important test for diagnosis of CPVT is the exercise stress test that induces ventricular arrhythmias in at least 80 % of CPVT patients. When the sinus rate exceeds an individual threshold rate (usually at 110–130 beats/min), VT typically appears. A diagnostic and highly reproducible marker of CPVT is the progressive worsening of arrhythmias during exercise. Intravenous infusion of catecholamines (e.g., isoproterenol or epinephrine) can also provoke progressive ventricular arrhythmias [2, 4]. After a first syncopal episode, if a high suspicion of CPVT exists, repeated exercise stress test is fundamental to avoid delay in the diagnosis [4]. Also, when a maximal exercise stress test is difficult, continuous ambulatory monitoring may reveal arrhythmias typical for CPVT. In some cases, an implantation of a loop recorder could be reasonable [1, 4]. Invasive EP testing is of no value in the diagnosis or risk stratification in patients with CPVT. Programmed electrical stimulation is not useful to arrhythmia induction [1, 3]. Genetic testing should be performed in all definitive CPVT probands (see Fig. 26.3).
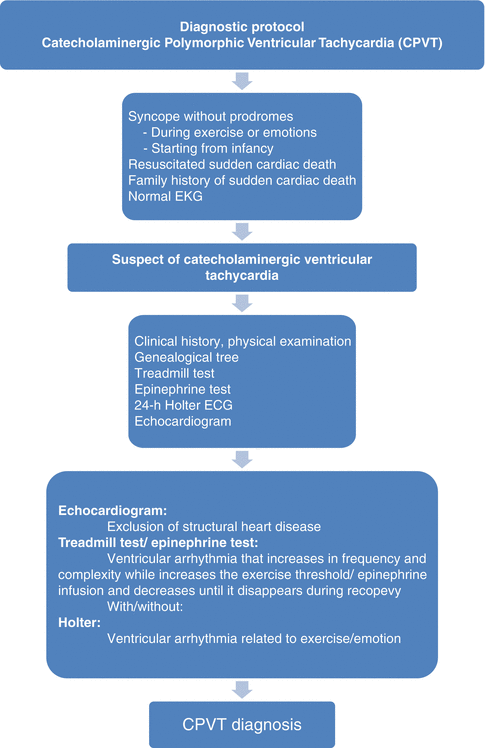
Fig. 26.3
Diagnostic protocol of catecholaminergic polymorphic ventricular tachycardia
Genetics of CPVT
Mutations in genes that encode for Ca2+ regulatory proteins are the principal cause of CPVT. The genetic variants of CPVT that have been described are the following: an autosomal dominant trait (CPVT1, most common) caused by mutations in the RyR2 gene and a recessive form (CPVT2, rare) associated with homozygous mutations in the CASQ2 gene [1].
RyR2 is the major calcium release channel of the sarcoplasmic reticulum, mediating excitation–contraction coupling. RyR2 mutations are present in approximately 50–70 % of patients with CPVT. Typical CPVT mutation of RyR2s shows gain-of-function defects following channel activation by PKA phosphorylation (in response to beta-adrenergic stimulation), leading to an uncontrolled Ca2+ release from the sarcoplasmic reticulum during electrical diastole that increases the diastolic membrane depolarizations (DADs) and triggers arrhythmias [2].
Another sarcoplasmic reticulum Ca2+ buffering protein associated with RyR2 is CASQ2 that plays an important role in the control of Ca2+ release from the sarcoplasmic reticulum to the cytosol. The hypothesis is that some of these mutations compromise CASQ2 synthesis and reduce expression or provoke complete absence of CASQ2 in the heart and others cause expression of defective CASQ2 proteins with abnormal regulation of cellular Ca2+ homeostasis [2, 3].
Mechanism of Ventricular Arrhythmias in CPVT
The central pathogenic abnormality in CPVT results from abnormalities in the control of sarcoplasmic reticulum Ca2+ release. The L-type Ca2+ channels in the cell membrane permit Ca2+ influx during the action potential plateau that triggers more Ca2+ release (Ca2+ transients) from the sarcoplasmic reticulum into the cytosol via activation of Ca2+ release channels (RyR2). This amplifying process, known as Ca2+-induced Ca2+ release, causes a quick raise in cytosolic Ca2+ concentration to an optimal level required for binding of Ca2+ to troponin C and induction of contraction [5]. During diastole, the sarcoplasmic/endoplasmic reticulum calcium adenosine triphosphatase (SERCA) resequesters most of the surplus Ca2+ in the cytosol into the sarcoplasmic reticulum, controlled by the phosphoprotein phospholamban. Additionally, to balance the Ca2+ that enters with the Ca2+ current, Na+–Ca2+ exchanger extrudes from the cell some of the Ca2+.
RyR2 mutations alter the physiological properties and function of RyR2 using some molecular mechanisms not fully understood. It has been suggested that the binding affinity of RyR2 for the regulatory protein calstabin-2 is reduced by CPVT mutations in RyR2. The closed conformational state of the RyR2 channel is stabilized by calstabin-2, which enables the channel to close completely during diastole (at low intracellular Ca2+ concentrations) and prevents aberrant Ca2+ leakage from the sarcoplasmic reticulum, ensuring muscle relaxation. The binding affinity of calstabin-2 is worsened by PKA phosphorylation (induced by beta-adrenergic stimulation) of the mutant RyR2 channels, increasing the probability of an open state at diastolic Ca2+ concentrations. So, diastolic Ca2+ leak from the sarcoplasmic reticulum during stress or exercise is the result of the incomplete closure of the mutant RyR2 channel during diastole [1, 6]. Other groups have suggested that in CPVT the mutated RyR2 channel is more sensitive to luminal (sarcoplasmic reticulum) Ca2+. So under baseline conditions, where sarcoplasmic reticulum load is normal, there is no Ca2+ leak, but under beta-adrenergic (sympathetic) stimulation, sarcoplasmic reticulum Ca2+ concentration raises above the reduced threshold, causing Ca2+ to leak out of the sarcoplasmic reticulum. A third hypothesis for RyR2-related CPVT is that the intermolecular interactions between discrete RyR2 domains necessary for proper folding of the channel and self-regulation of channel gating are impaired by the mutations in RyR2 [6].
The most important Ca2+ storage protein in the sarcoplasmic reticulum is calsequestrin (CASQ2) and forms a part of a quaternary complex with RyR2, triadin, and junction, which play a major role in regulating intracellular Ca2+. Calsequestrin, as a high-capacity, low-affinity Ca2+-binding protein, is able to bind luminal Ca2+ during diastole, buffering Ca2+ within the sarcoplasmic reticulum and preventing diastolic Ca2+ release via RyR2 to the cytosol. For effective termination of sarcoplasmic reticulum Ca2+ release and prevention of spontaneous Ca2+ release during diastole, the control of RyR2s by luminal Ca2+ is required. CASQ2 mutations lead to diminished Ca2+ signaling refractoriness and generation of arrhythmogenic spontaneous Ca2+ releases [6]. The main arrhythmogenic mechanism in CPVT is DADs and triggered activity because the bidirectional ECG pattern of this VT corresponds to the arrhythmias related with intracellular Ca2+ overload and the DADs documented during digitalis toxicity. Mutations in RyR2 or CASQ2 lead to the activation of the Na+–Ca2+ exchanger enhanced by cytosolic Ca2+ overload. This mechanism generates a net inward current (the so-called transient inward current) that provokes DADs and if they reach the threshold for Na+ channel activation can trigger abnormal beats [1, 3]. Low-amplitude DADs usually are not clinically significant. A decrease in the initiating cycle length probably is the most important factor that leads subthreshold DADs to reach threshold; indeed fast heart rates increase both the amplitude and rate of the DADs. Moreover, catecholamines can facilitate the development of DADs because the stimulation of beta-adrenergic receptors and subsequently cAMP increases the L-type Ca2+ current and leads to an increase in transsarcolemmal Ca2+ influx and intracellular Ca2+ overload; also the enhancement of the activity of the Na+–Ca2+ exchanger increases the likelihood of DAD-mediated triggered activity; finally, the enhancement of the uptake of Ca2+ by the sarcoplasmic reticulum leads to a greater amount of Ca2+ stored in the sarcoplasmic reticulum and the subsequent major release of Ca2+ from the sarcoplasmic reticulum during contraction. The increased susceptibility to ventricular arrhythmias in CPVT patients during exercise and emotional stress is associated with increased sympathetic stimulation and higher heart rates.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


