Figure 31–1.
RyR2 mutational clusters. Clusters with frequent mutations are depicted with their location along RyR2 amino acid (AA) sequence. Percentage of known (published) mutation foe each cluster is also reported. Mutations outside the canonical clusters are depicted as dots in the figure. Each dot represents a unique mutation. Green dots represent published mutation while black dots represent unpublished mutation form our database. MCL Molecular Cardiology Laboratories, Pavia, Italy
According to a recent consensus document [12], only the analysis of RyR2 is recommended on a routine basis. CASQ2 screening is indicated in the presence of possible recessive inheritance and also in sporadic CPVT RyR2-negative patients since compound heterozygosity is possible [13].
Pathophysiology
RyR2 channels are normally activated by Ca2+ entering the cell through the voltage-dependent L-type Ca2+ channels, causing the release of Ca2+ from the sarcoplasmic reticulum (SR) into the cytosol, a mechanism known as Ca2+-induced Ca2+ release (CICR) (Fig. 31.2) [14]. The hypothesis that arrhythmias in CPVT are initiated by delayed afterdepolarizations (DADs) and triggered activity had been advanced since the identification of the first RyR2 mutations. The hypothesis was proven correct with the availability of the conditional knock-in mouse model carrier of the RyR2R4496C+/− that develops the typical bidirectional VT [15] and DADs/triggered activity (Fig. 31.3) [16].
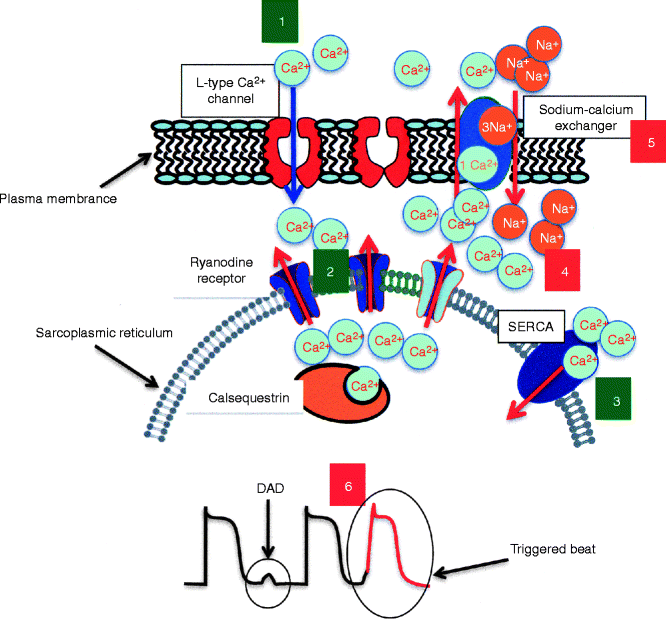
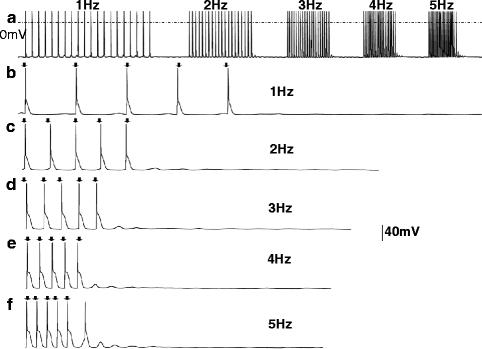
Figure 31–2.
Cartoon illustrating the steps of calcium induced calcium release process (green numbers) and the cellular pathophysiology of CPVT (red numbers). (1) Calcium ions enter the cell through L-type channels during phase 1–2 of the action potential and trigger the release of a large amount of calcium form the sarcoplasmic reticulum (2) which allows contraction. After calcium is freed form actin-myosin complex it is transported back to the sarcoplasmic reticulum by the SERCA pump (3). When diastolic calcium leak occurs due to RyR2 or CASQ2 mutations the cell must get rid of the cytosolic overload (4) and the sodium-calcium exchanger is activated (5). The exchanger removes one calcium ion every three sodium ions entering the cell. Therefore it generates a depolarizing current (transient inward current) which underlies delayed afterdepolarizations (DAD); if large enough, DADs can trigger an action potential (6)
Figure 31–3.
(a) Action potentials recorded from a RyR2R4496C+/− myocyte stimulated at 1–5 Hz are shown: DADs develop when pacing is interrupted. (Panels b–f) The last five driven action potentials at each pacing frequency and DADs and triggered activity developing when pacing is discontinued are shown. Note that DAD amplitude increases and DAD coupling interval decreases at faster pacing frequencies. At 5 Hz, (panel f) one triggered beat develops
Pathophysiology of RyR2 Mutations
Jiang et al. [17, 18] directly determined the impact of a number of CPVT RyR2 mutations on the sensitivity of single RyR2 channels to luminal Ca2+ activation using single channel recordings in planar lipid bilayers. In vitro expression of many RyR2 mutations showed a consistent behavior characterized by an enhanced response of the channel to luminal Ca2+ activation while only few of them seem to affect the response of the RyR2 channel to cytosolic Ca2+, or both cytosolic and luminal sensitivity [19].
The increased sensitivity is the cause of a reduced threshold for the so-called Store Overload Induced Calcium Release (SOICR). In a nutshell the macroscopic effect of mutations is that of lowering the threshold for spontaneous calcium release from the sarcoplasmic reticulum. Thus, upon adrenergic activation (which physiologically increases SR calcium content), it becomes easier for SR calcium release to occur [20] and to generate delayed afterdepolarization and triggered activity during electrical diastole (Fig. 31.4) [21].
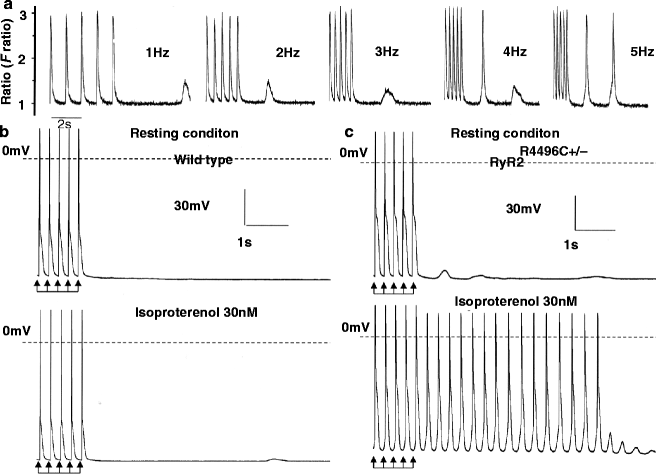
Figure 31–4.
(a) A Fura-2 loaded RyR2R4496C+/− myocyte in presence of isoproterenol (30 nM), spontaneous Ca2+ release showed positive frequency dependent manner. Action potential recording from a WT myocyte (panel b) and a RyR2R4496C+/− myocyte (panel c) in the absence (top panel) and in the presence (bottom panel) of isoproterenol (30 nM). Arrows indicate the last five paced action potentials
The molecular mechanisms for such calcium sensitization are complex and still incompletely understood. Two major hypotheses have been proposed: (A) defective interaction between protein domains controlling channel gating (unzipping) or (B) the unbinding of FKBP12.6 that is believed to be a RyR2 stabilizer [22–24]. This latter hypothesis is questioned by the evidence that K201, a 1,4-benzothiazepine derivative (formerly called JTV 519) which enhances the binding of FKBP12.6 to RyR2, is unable to prevent triggered activity and arrhythmias in RyR2R4496C+/− mice [16]. More recently Guo et al. reported that 10–20 % of endogenous myocyte RyR2s have FKBP12.6 associated and PKA-dependent RyR2 phosphorylation has no significant effect on binding of either FKBP12 or 12.6 to RyR2 in myocytes [25]. Those data suggested that FKBP12.6 would not be the key point for the arrhythmogenesis of CPVT.
It is known that Purkinje fibers are more susceptible to Ca2+ overload than ventricular muscle possibly because of their greater sodium load and longer action potential duration. Recently two independent groups reported that Purkinje fibers isolated from RyR2R4496C+/− mice display a greater propensity to develop intracellular Ca2+ handling disorder than ventricular myocytes, suggesting that focally activated arrhythmias might originate in the specialized electrical conducting cells of the His-Purkinje system in CPVT [26, 27]. More interestingly epicardial mapping study in RyR2R4496C+/− hearts revealed that the bidirectional morphology of VT is caused by alternating firing of triggered activity from the right and left bundle [28].
Pathophysiology of CASQ2 Mutations
The CASQ2 protein serves as the major Ca2+ reservoir within the SR and it is part of a protein complex that contains the ryanodine receptor. In the presence of 50 % loss of calsequestrin (in heterozygous carriers) a normal clinical phenotype is observed; thus suggesting that the heart can tolerate quite severe CASQ2 reductions. It has been demonstrated that the underlying mechanism for the CASQ2-CPVT is associated with impaired SR Ca2+ buffer and defects of CASQ2-dependent regulation of RyR2 channel open probability. More recently various transgenic mouse models harboring CASQ2 mutations have been developed [29–31]. Those mouse models exhibit severe CPVT phenotype and demonstrate marked-to-complete reduction of CASQ2 protein levels [29, 30]. Therefore reduced CASQ2 expression appears to represent a common feature of autosomal recessive CPVT. Interestingly this effect may induce a number of abnormal adaptive responses, such as increasing RyR2 and calreticulin, decreasing triadin and junction [29–31]. More interestingly, despite lack of cardiac hypertrophy and fibrosis in youth mice, electron microscopy has revealed ultrastructural remodeling in SR, including enlarged SR volume and widened jSR dimension, suggesting ultrastructural cardiomyopathy for autosomal recessive CPVT [29, 31]. Isolated ventricular myocytes with CASQ2 mutation present DAD as well as early afterdepolarization due to abnormal Ca2+ leak from RyR2 [29]. The pathophysiology of CPVT is schematically represented in Fig. 31.3.
Clinical Management
Clinical Manifestations
Syncope, triggered by exercise or emotional stress, is often the first manifestation of CPVT [3, 10]. These symptoms are accompanied by family history of sudden death in approximately 30 % of patients. The mean age at first event is between was 7 and 12 years [3, 10, 32]. Increasing evidences show that sudden cardiac death can be the first manifestation of the disease [10, 11] and that CPVT may be a cause of sudden infant death syndrome [33] and idiopathic ventricular fibrillation [34].
Electrocardiogram
The resting ECG of CPVT patients is normal with the exception of prominent U waves and mild sinus bradycardia in some patients [3, 32]; the CPVT phenotype manifests preferentially upon adrenergic stimulation with the onset of a reproducible pattern of ventricular arrhythmias. Exercise stress test usually elicits isolated atrial and ventricular extrasystoles at heart rate between 95 and 110 beats per minute [3, 10]. The complexity and frequency of ventricular arrhythmia progressively worsen with increase of workload (Fig. 31.5). The typical ventricular tachycardia (VT) observed in CPVT is the so-called bidirectional VT: an alternating QRS axis morphology with a rotation of 180° on a beat-to-beat basis.
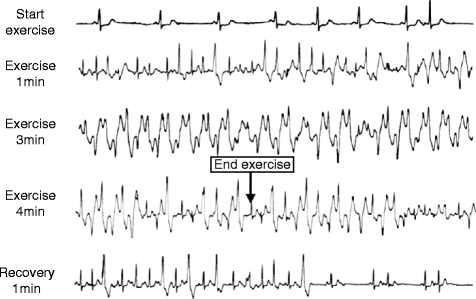
Figure 31–5.
Exercise stress test in a patient with polymorphic VT and RyR2 mutation. Ventricular arrhythmias are observed with a progressive worsening during exercise. Typical bidirectional VT develops after 3 min of exercise with a sinus heart rate of approximately 120 bpm. Arrhythmias rapidly recede during recovery
Diagnosis
Exercise or emotion-induced syncope in a patient with a normal electrocardiogram (normal QT interval) and without structural abnormalities, should always prompt the suspect of CPVT. Exercise stress test is the most important tool for diagnosis since the bidirectional or polymorphic VT may be reproducibly elicited during physical activity in most of the patients. In patients showing polymorphic (and not bidirectional) VT, the progressive worsening of arrhythmias with exercise is to be considered as diagnostic for CPVT. Holter monitoring can also be useful especially in young children in whom it might be difficult to reach high heart rate during treadmill test or for patients particularly sensitive to emotional stress.
Interestingly, the bidirectional VT is a common finding in patients with in Andersen syndrome [35]. This disease is a variant of the Long QT syndrome (also called LQT7) caused by loss of function mutations of the KCNJ2 gene encoding for the cardiac inward rectifier potassium current. The reasons for this apparent “phenocopy arrhythmia” are still unclear but differential diagnosis between the two conditions should be considered and it has clinical relevance since KCNJ2 mutation are often benign and only exceptional cause of sudden cardiac death.
Treatment
Chronic treatment with beta-adrenergic blockers can prevent recurrent syncope in the majority of CPVT patients (Fig. 31.6). Nadolol at a daily dose of 1–2.5 mg/kg/day is the drug used at our center but propranolol can also be considered. Less experience is available with other beta blockers; in our experience metoprolol and atenolol are less effective due to their weaker effect in blunting exercise-induced heart rate increase. It is also important to be aware that dosage should be always individualized to achieve the best possible control of arrhythmias during exercise stress testing. Individual tolerability of the drug should be always considered. In our experience, some patients may be treated with up to 3.5–4 mg/kg/day of nadolol without clinically relevant side effects and increased protection from arrhythmic events. However, it is important to be aware that some patients still develop repetitive ventricular arrhythmias during exercise stress test even with maximal tolerated dose of beta blocking agents (see below).
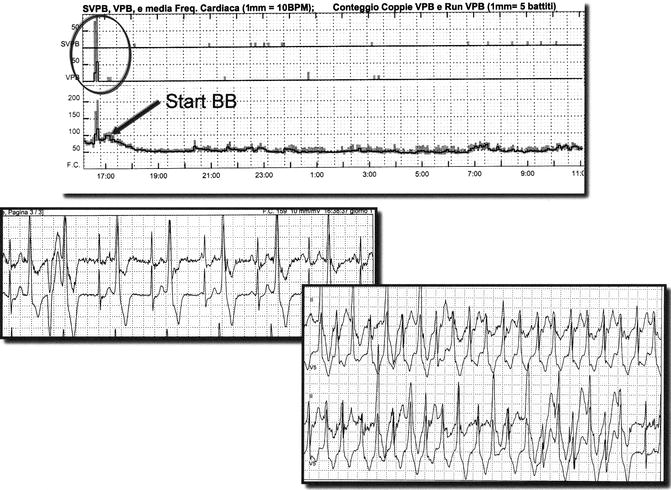
Figure 31–6.




Holter monitoring and beta blocker therapy in CPVT. The upper panel shows the trend of arrhythmias and heart rate. When beta blocker therapy (nadolol 1.5 mg/kg) is started (arrow) hear rate decreases and the incidence of arrhythmias dramatically decreases. Lower panels show ventricular bigeminy and ventricular tachycardia recorded in the first hour before beta blockers
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
