Case 25
HISTORY AND PHYSICAL EXAMINATION
| Right | Left | |
|---|---|---|
| Shoulder abduction | 2/5 | 5/5 |
| Elbow flexion | 3/5 | 5/5 |
| Elbow extension | 4−/5 | 5/5 |
| Pronation | 0/5 | 3/5 |
| Fingers flexion | 0/5 | 3/5 |
| Wrist flexion | 1/5 | 1/5 |
| Wrist extension | 2/5 | 5/5 |
| Finger extension | 3/5 | 4−/5 |
| Finger abduction | 4−/5 | 3/5 |
| Right | Left | |
|---|---|---|
| Hip flexion | 5/5 | 5/5 |
| Hip extension | 5/5 | 5/5 |
| Knee extension | 5/5 | 5/5 |
| Knee flexion | 5/5 | 5/5 |
| Foot dorsiflexion | 5/5 | 1/5 |
| Toe dorsiflexion | 5/5 | 0/5 |
| Plantar flexion | 5/5 | 5/5 |
| Ankle inversion | 5/5 | 5/5 |
| Ankle eversion | 5/5 | 1/5 |
Please now review the Nerve Conduction Studies and Needle EMG tables.
QUESTIONS
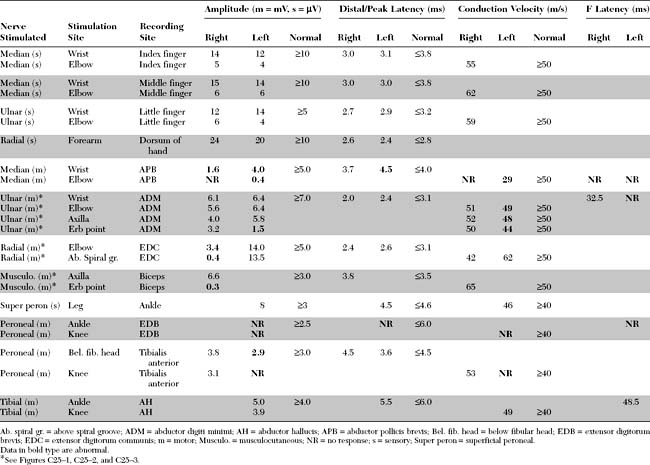
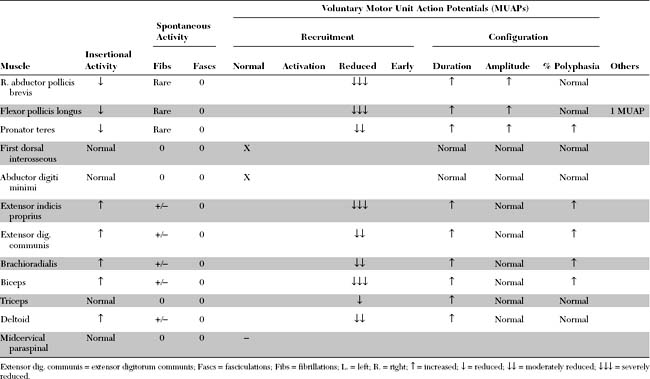
EDX FINDINGS AND INTERPRETATION OF DATA
Relevant EDX findings in this case include:
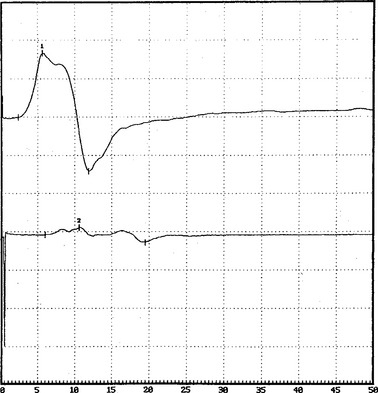
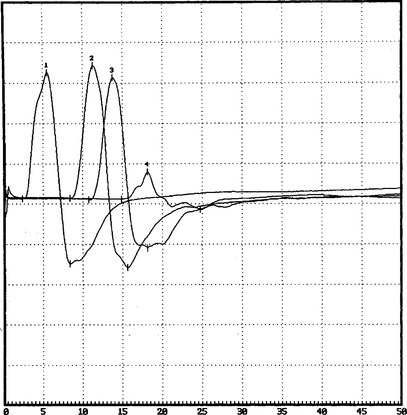
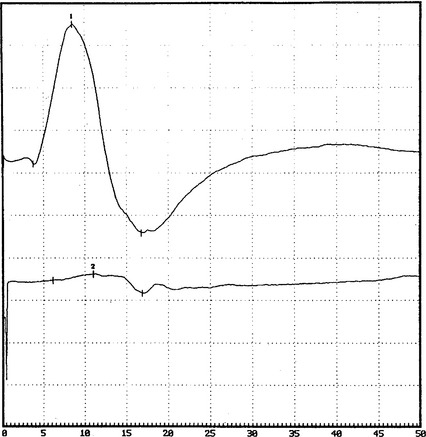
DISCUSSION
Definition and Pathogenesis
Multifocal motor neuropathy (MMN), described in the mid-1980s, is a rare disorder with a prevalence of 1 to 2 individuals per 100 000. It is characterized by specific EDX finding, i.e., motor conduction blocks, which is the gold standard for diagnosis. The disorder is important to recognize since it is treatable and responsive to immunomodulating therapies, and may mimic amyotrophic lateral sclerosis (ALS) which has a poor prognosis for survival.
CLINICAL FEATURES
Multifocal motor neuropathy presents insidiously with asymmetrical weakness often in the distribution of individual nerves. The age of onset of first symptoms is between 20 and 50 years of age in about 80% of patients, and the disorder is more common in men than women (ratio of 2.6/1). In more than 80% of patients, the weakness starts in the upper limbs, usually hand and forearm muscles. Other than the hypoglossal nerve, cranial nerve involvement is rare. Unilateral or bilateral phrenic nerve palsy causing respiratory failure may occur and is occasionally the presenting symptom. The disorder is slowly progressive, usually for more than 6 months and often years. Sometimes, the history is one of a stepwise progression with episodes of rapid worsening followed by prolonged periods of stabilization. The deep tendon reflexes are variable; they are usually depressed or absent diffusely or in weak limbs only. They may be normal or even brisk in one-third of patients, leading to confusion with ALS. Muscle atrophy is not prominent in weak muscles, despite the degree and chronicity of weakness; it may be present over the long term in the distribution of one or more affected nerves, implicating motor axon loss and predicting poor response to therapy. Mild sensory complaints may be present, but the sensory examination is usually normal except for minor vibration sense abnormalities in the lower extremities. A high titer of anti-GM1 antibody is present in approximately 50% of patients, although this varies between 30 and 80%, probably due to the different methodology utilized for antibody measurement. The cerebrospinal fluid protein is usually normal, but may be elevated in one-third of patients without exceeding 100 mg/dL.
Multifocal motor neuropathy should be distinguished from amyotrophic lateral sclerosis, particularly in patients with predominant or exclusive lower motor neuron findings. Clues on clinical examination of patients with MMN include the distribution of weakness, which follows peripheral nerves rather than spinal segments, the insidious course over many years, and the lack of pyramidal signs. It should be cautioned that preserved or brisk reflexes may be present in one-third of patients with MMN. Also, other forms of anterior horn cell disorders, such the spinal muscular atrophies, brachial amyotrophic diplegia (the flail arm syndrome) and monomelic amyotrophy (Hirayama disease) should be excluded. The flail arm syndrome (brachial amyotrophic diplegia), a variant of the progressive muscular atrophy form of ALS, is characterized by progressive proximal and distal upper limb weakness and ultimate variable involvement of the lower limbs. Monomelic amyotrophy (Hirayama disease) affects young men between the age of 15 and 22 and presents with an asymmetrical wasting and weakness of distal upper limb muscles. The disorder is benign, initially progressive over several years and then becoming static. Finally, MMN should be distinguished from other chronic acquired demyelinating peripheral polyneuropathies that may be associated with conduction block or predominantly motor including chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) and its variant the Lewis-Sumner syndrome (multifocal acquired demyelinating sensory and motor neuropathy, MADSAM), osteosclerotic myeloma (POEMS syndrome), and MGUS neuropathy.
Diagnostic criteria for MMN were proposed. The aim of these criteria is to strengthen the diagnosis of MMN and exclude other disorders that may mimic it. They mostly emphasize the mononeuropathy multiplex-like distribution of weakness, presence of multifocal motor conduction block, lack of sensory loss and lack of pyramidal signs. Table C25-1 shows recently accepted criteria for accurate diagnosis of MMN.
Table C25-1 Criteria for the Diagnosis of Multifocal Motor Neuropathy



