Case 20
HISTORY AND PHYSICAL EXAMINATION
| Right | Left | |
|---|---|---|
| Deltoid | 4/5 | 4/5 |
| Triceps | 4/5 | 4/5 |
| Biceps | 4/5 | 4/5 |
| Hand grip | 4+/5 | 4+/5 |
| Iliopsoas | 4−/5 | 4−/5 |
| Quadriceps | 4−/5 | 4−/5 |
| Hamstrings | 4−/5 | 4−/5 |
| Ankle dorsiflexion | 4+/5 | 4+/5 |
| Plantar dorsiflexion | 4+/5 | 4+/5 |
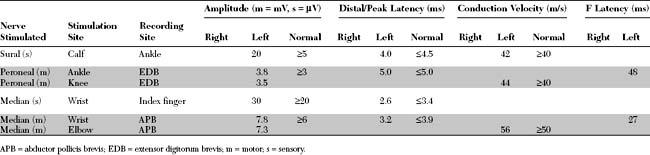
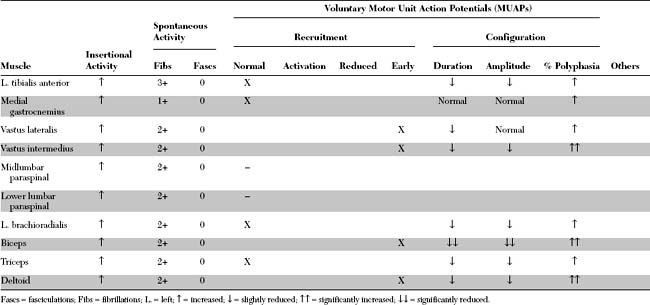
An electrodiagnostic (EDX) examination was performed.
Please now review the Nerve Conduction Studies and Needle EMG tables.
QUESTIONS
EDX FINDINGS AND INTERPRETATION OF DATA
Pertinent EDX findings in this case include the following:
DISCUSSION
Classification and Pathology
The inflammatory myopathies are a heterogeneous group of disorders that share a common pathologic feature: inflammatory cells in muscles. They comprise three major categories of muscle disease, polymyositis (PM), dermatomyositis (DM), and inclusion body myositis (IBM), conditions that are clinically, histologically, and pathogenetically distinct. Inflammatory myopathies are often classified into two major types (Table C20-1), a primary and secondary (associated with systemic or other identifiable disorders).
Table C20-1 General Classification of Inflammatory Myopathies
With connective tissue diseases (overlap syndromes), e.g., scleroderma, systemic lupus erythematosus |
Pathologically, the inflammatory myopathies are characterized by mononuclear cell inflammation, segmental muscle fiber necrosis, and muscle fiber regeneration. The pathologic findings in PM and DM have both common and diverse features. Both also are different from the findings in inclusion body myositis. Table C20-2 shows the major differences among these three primary inflammatory myopathies.

Clinical Features
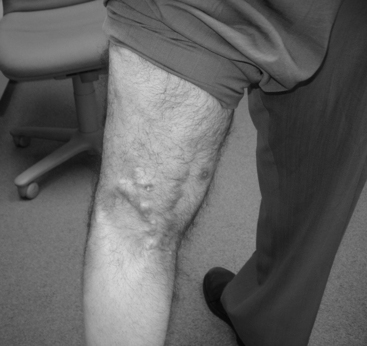
In DM only, there is an associated skin rash that can be the presenting symptom. Two classic rashes are characteristic. The first is a “heliotrope rash,” an erythematous, violaceous (hence the name) rash over the malar and periorbital areas that may extend to involve other sun-exposed areas, such as the dorsum of the hands, knees, elbows, or forehead. The second rash is Gottron papules, an erythematous papular rash over the knuckles of the fingers. Subcutaneous calcinosis complicates up to half of children with juvenile DM and may be present in chronic DM. These lesions are most often seen in the buttocks, thighs, knuckles, and elbows (Figure C20-1). They may be painful, ulcerate through the skin, or get infected.
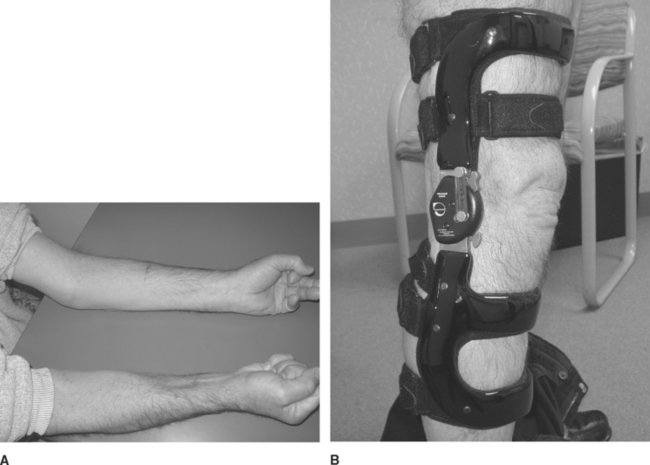
In contrast to PM and DM, IBM affects patients over the age of 50, has a male predominance, and is more common in the white than black population. The onset is insidious and it progresses slowly, evolving over years. It is the most common inflammatory myopathy in patients over the age of 50. Clinically, IBM has a unique muscle involvement that easily distinguishes it from the other inflammatory myopathies. There is usually asymmetrical involvement of the finger flexors and wrist flexors in the upper extremities, and the knee extensors and ankle dorsiflexors in the lower extremities (Figure C20-2). Dysphagia is common and afflicts up to 60% of patients as the disease progresses.
The diagnoses of DM and PM are confirmed based on the combination of clinical, laboratory, electrophysiologic, and pathologic findings. In 1975, Bohan and Peter proposed a classification that has been used since, with some revisions, in confirming the diagnoses of PM and DM. Based on these criteria, the confidence limits in the diagnosis of PM or DM range from definite to probable to possible (Table C20-3). The differential diagnoses of PM include IBM, polymyalgia rheumatica, metabolic myopathies (such as acid maltase deficiency), and limb girdle muscular dystrophy. In dermatomyositis, the presence of typical skin rash combined with muscle weakness often is diagnostic.
Table C20-3 Criteria and Confidence Limits in the Diagnosis of Polymyositis and Dermatomyositis
| Criteria | |
| Predominantly proximal (limb girdle and neck flexor muscles), usually symmetrical, muscle weakness progressing over weeks or months, with or without myalgia, dysphagia or respiratory muscle involvement | |
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

| |