Case 17
HISTORY AND PHYSICAL EXAMINATION
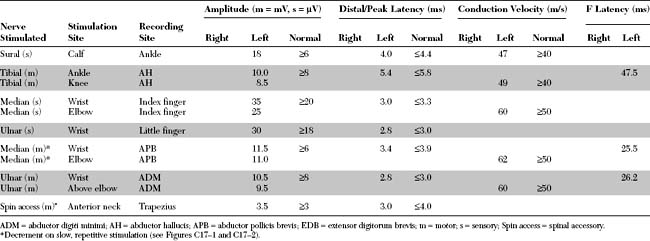
An electrodiagnostic (EDX) examination was performed.
Please now review the Nerve Conduction Studies and Needle EMG tables.
QUESTIONS
EDX FINDINGS AND INTERPRETATION OF DATA
Pertinent EDX findings include the following:
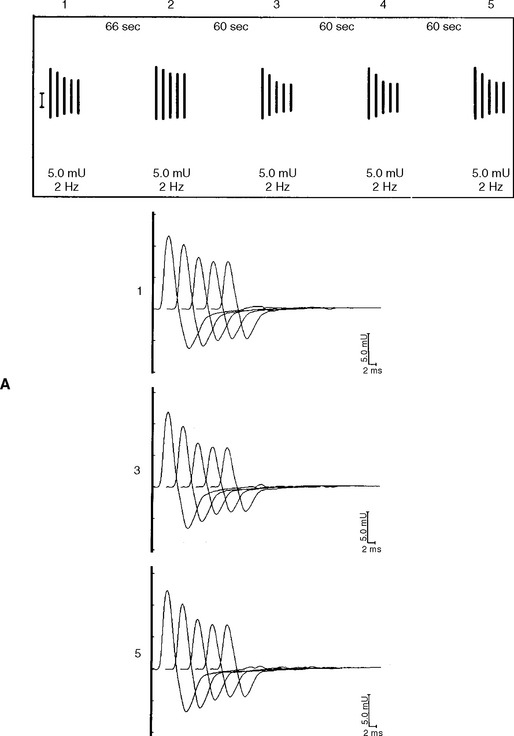
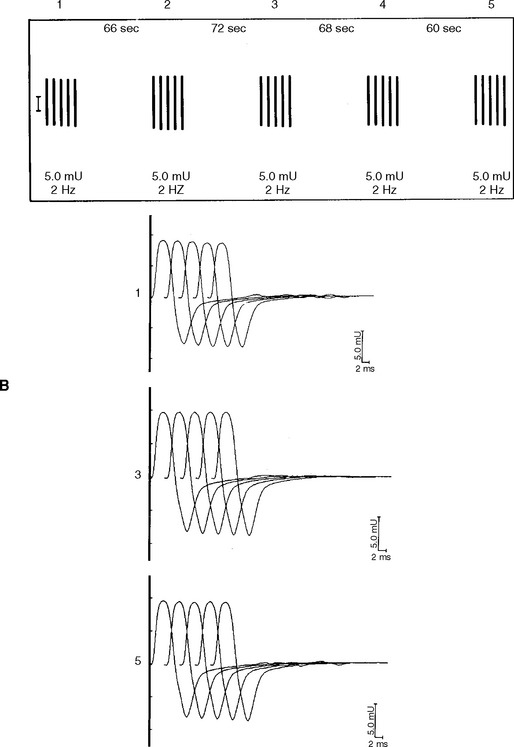
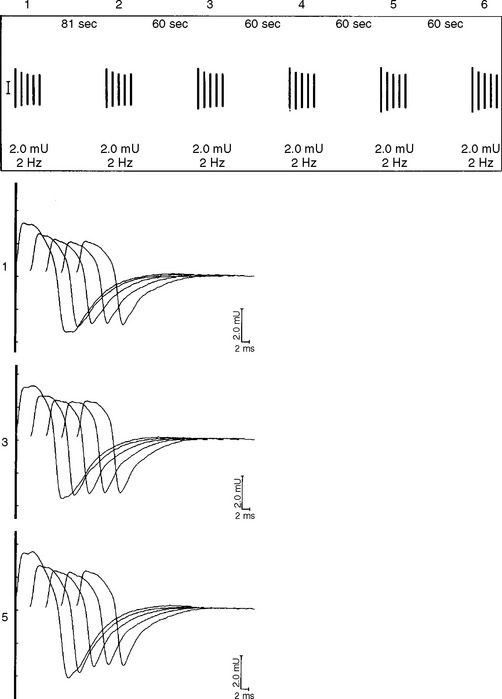
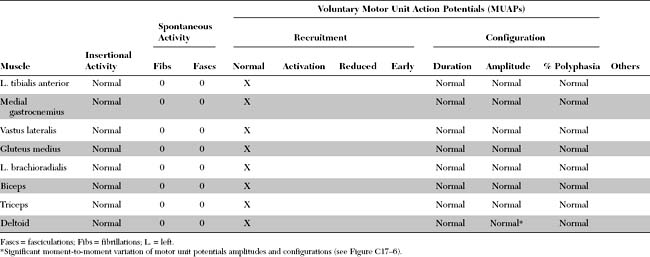
DISCUSSION
Anatomy and Physiology
Neuromuscular Junction
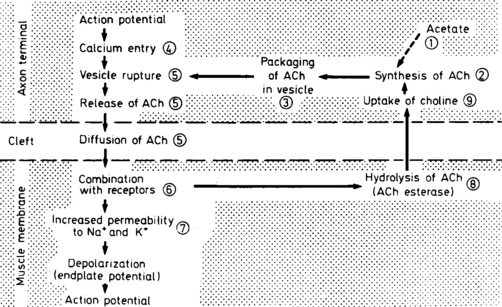
The neuromuscular junction (NMJ) is the site where the motor neuron makes contact with the skeletal muscle fiber’s membrane (sarcolemma). It is near the center of the muscle fiber where there is a cup-shaped depression of the sarcolemma, called the endplate. The NMJ is a chemical synapse that is essential for transmitting action potentials from the terminal nerve branches to muscle fibers. This synapse utilizes acetylcholine (ACH) as a transmitter that binds to specific receptors in the junctional membrane, resulting local depolarizations that spread and trigger all-or-none muscle action potential. The NMJ is divided into a presynaptic terminal, a synaptic cleft, and a postsynaptic region (Figure C17-3).
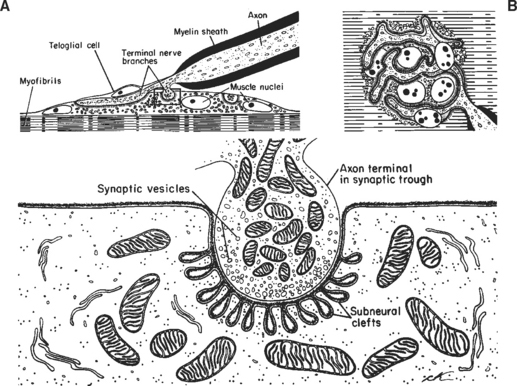
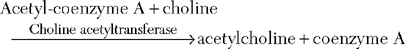
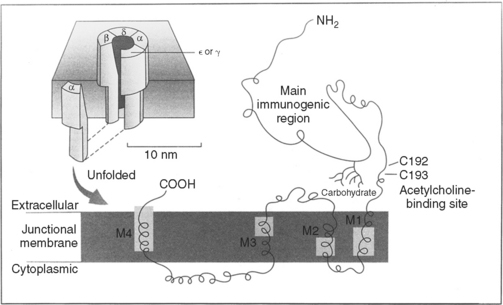
Neuromuscular Transmission
Clinical Features
Myasthenia gravis (MG) is the best understood and most thoroughly studied of all human organ-specific autoimmune diseases. It is characterized by a reduction of skeletal muscle postsynaptic ACH receptors resulting in a decrease in the EPP necessary for action potential generation. In the majority of patients, MG is caused by an antibody-mediated attack on the postsynaptic nicotinic ACH receptors in the neuromuscular junction. In a small number of patients, other antigenic targets, such as the muscle specific tyrosine kinase (MuSK), may exist. Myoid cells and other stem cells within the thymus gland, which is hyperplastic in at least two-thirds of patients with MG, may serve as autoantigens by expressing on their surface the ACH receptor or one of its protein components.
The hallmarks of MG are muscle weakness and fatigability. The symptoms are intermittent and are usually worse with activity and improve after rest. Generally, patients are much better in the morning than in the evening. Ocular symptoms (diplopia and/or ptosis) are extremely common and are the presenting signs in more than one half of patients. Most importantly, almost all patients at some point during the course of their illness develop ocular manifestations. Also, the disorder continues to be restricted to the extraocular muscles in 15% of patients, hence the designation ocular myasthenia. Additionally, only 3–10% of patients with ocular myasthenia generalize if no other symptoms appear after three years from initial presentation. Bulbar muscle weakness is the initial presenting manifestation in about 20% of patients and is seen in over 30% of patients during the course of their disease. Bulbar weakness is a major contributor to disability throughout the course of the disease. It manifests as dysarthria, nasal speech, dysphagia, chewing difficulties, or nasal regurgitation. Occasionally, the jaw muscle weakness may be severe leading to a “jaw drop” and patients often hold their jaw closed, a highly pathognomonic manifestation of MG. Limb weakness, mostly of proximal muscles, is seen as the initial symptom in 20% of patients. At times, the generalized weakness is severe and involves the respiratory muscles, resulting in respiratory failure that requires mechanical ventilation, a situation often referred to as myasthenic crisis. Because of variable clinical severity, MG is usually classified into five main categories (Table C17-1).
Table C17-1 Myasthenia Gravis Foundation of America Clinical Classification
| Class I | Any ocular muscle weakness |
| May have weakness of eye closure | |
| All other muscle strength is normal | |
| Class II | Mild weakness affecting other than ocular muscles |
| May also have ocular muscle weakness of any severity | |
| IIa | Predominantly affecting limb, axial muscles or both |
| May also have lesser involvement of oropharyngeal muscles | |
| IIb | Predominantly affecting oropharyngeal, respiratory muscles, or both |
| May also have lesser involvement of limb, axial muscles, or both | |
| Class III | Moderate weakness affecting other than ocular muscles |
| May also have ocular muscle weakness of any severity | |
| IIIa | Predominantly affecting limb, axial muscles or both |
| May also have lesser involvement of oropharyngeal muscles | |
| IIIb | Predominantly affecting oropharyngeal, respiratory muscles, or both |
| May also have lesser involvement of limb, axial muscles, or both | |
| Class IV | Severe weakness affecting other than ocular muscles May also have ocular muscle weakness of any severity |
| IVa | Predominantly affecting limb, axial muscles or both May also have lesser involvement of oropharyngeal muscles |
| IVb | Predominantly affecting oropharyngeal, respiratory muscles, or both |
| May also have lesser involvement of limb, axial muscles, or both | |
| Class V | Defined by intubation, with or without mechanical ventilation, except when employed during routine postoperative management. The use of a feeding tube without intubation places the patient in class IVb |
The diagnosis of MG may be made on clinical grounds, especially when reproducible fatigability of eyelids or extraocular muscles is confirmed. However, laboratory testing is frequently needed, and recommended, for confirmation (Table C17-2):
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


