Chapter 3 Cardiovascular Pharmacology
In this chapter the pharmacology of cardiovascular drugs that are used in the intensive care unit (ICU) is reviewed. Specific indications for particular drugs are discussed in other relevant chapters. Guidelines for the reintroduction of medications following routine cardiac surgery are provided in Chapter 17.
INOTROPES AND VASOPRESSORS
Infusions of vasoactive drugs are prescribed in different ways in different institutions. Three common methods are micrograms per kilogram per minute (μg/kg/min), micrograms per minute (μg/min), and milligrams per hour (mg/hr). In this book μg/kg/min is used. A conversion among the methods is provided in Appendix 1.
Sympathomimetics
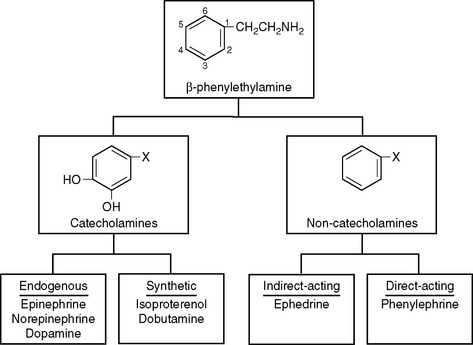
All sympathomimetics are derived from β-phenylethylamine. The presence of hydroxyl groups on the 3- and 4-carbons in the benzene ring designates a compound as a catecholamine, which may be endogenous or synthetic (Fig. 3-1). The noncatecholamine sympathomimetics include a diverse range of drugs, such as the asthma medication albuterol and the central nervous system stimulant amphetamine. Two commonly used vasoactive noncatecholamine sympathomimetics are ephedrine and phenylephrine.
Mechanism of Action
Sympathomimetics bind to and stimulate adrenergic receptors that are located on cell membranes. In 1948, Alquist described two adrenergic receptor subtypes, alpha (α) and beta (β), based on their relative responsiveness to norepinephrine, epinephrine, and isoproterenol.1 In the 1970s this classification was refined to include α1, α2, β1, and β2 receptor subtypes. Subsequently, further divisions of each receptor subtype have been discovered, but clinically useful drugs to exploit these expanded classifications have not been developed.
Adrenergic receptors are part of a family of receptors known as G protein coupled receptors. Receptor stimulation by an agonist (see Chapter 4) facilitates the binding of the nucleotide guanosine triphosphate to a G protein, which activates it. The activated G protein then stimulates or inhibits one of a number of second messenger systems. Two second messenger systems mediate the actions of adrenergic receptors:
Receptor Effects
The direct cardiovascular effects of the adrenergic receptor subtypes are summarized in Table 3-1.
Table 3-1 Effects of Adrenergic Receptor Subtypes on the Cardiovascular System
| Receptor | Location | Action |
|---|---|---|
| α1 | Vasoconstriction | |
| α2 | Vasodilation | |
| β1 | Heart | |
| β2 | Veins | Vasodilation |
β Receptors.
β2 Receptors are found on the heart and throughout the vasculature, particularly the arterioles of skeletal muscle, the coronary circulation, and the liver. Their stimulation leads to vasodilation and enhanced diastolic relaxation (lusitropy). Outside of the cardiovascular system, β2 receptor activation causes bronchodilation, uterine and bladder relaxation, and decreased gastrointestinal motility.
Individual Sympathomimetics
Norepinephrine.
Norepinephrine causes potent stimulation at α and β1 receptors, but unlike epinephrine, it has minimal effect at β2 receptors. Blood pressure is reliably increased but the effect on cardiac output is variable. Although β1 receptor stimulation has a direct inotropic effect, in the setting of hypovolemia or impaired ventricular function, increased left ventricular afterload due to α1 receptor stimulation can cause cardiac output to fall. Similarly, the effect on heart rate is variable: direct β1 stimulation has a chronotropic effect but increased blood pressure can cause baroreceptor-mediated bradycardia.
Norepinephrine is useful following cardiac surgery to counter the vasodilatory effects of cardiopulmonary bypass and sedation. However, doses above 0.05 to 0.1 μg/kg/min should be avoided in patients with impaired ventricular function unless cardiac output is being measured. Norepinephrine is commonly combined with an inodilator such as dobutamine or milrinone. Norepinephrine is typically commenced at a dose of 0.01 to 0.05 μg/kg/min and titrated to blood pressure. There is no maximum dose, but infusions greater than 0.1 to 0.2 μg/kg/min are rarely needed in cardiac surgery patients except in the presence of vasoplegic syndrome (Chapter 2) or septic shock, in which case doses as high as 0.5 to 1 μg/kg/min may be required. Troublesome metabolic effects, particularly lactic acidosis, are much less common with norepinephrine than with epinephrine.2
Dopamine.
Dopamine is a precursor to norepinephrine and is itself an important neurotransmitter in the peripheral and central nervous systems. Dopamine stimulates α and β receptors and type 1 and 2 dopamine (DA) receptors. DA-1 receptors are found in the renal, mesenteric, and cerebral circulations,3 and their stimulation results in vasodilation. DA-1 receptors are also found in the renal tubule, where they mediate natriuresis. DA-2 receptors are analogous to α2 receptors in that they are found presynaptically and inhibit the release of norepinephrine. Dopamine also has an indirect mechanism of action.
At low doses (<3 μg/kg/min), dopaminergic effects predominate. At higher doses, initially β receptor effects predominate; then α receptor effects predominate. The widely accepted dose range is 3 to 10 μg/kg/min for β effects and more than 10 μg/kg/min for α effects. However, these dose ranges must be viewed with skepticism. There is huge individual variability in the pharmacokinetics of dopamine such that dramatically different plasma concentrations may occur in different patients who are receiving the same dose.4 Furthermore, the clinical effects of a given plasma concentration are dependent on the functional activity of the adrenergic receptors. β Receptors are desensitized in a variety of clinical settings, including after cardiac surgery and with heart failure.5–7 Because of its indirect action, dopamine has reduced effectiveness in patients with heart failure or shock. Despite these caveats, it is generally true that as the dose of dopamine increases, there is a progressive increase in blood pressure and heart rate.
Dopamine at a dose of 1 to 3 μg/kg/min has been termed “renal-dose dopamine” and has traditionally been used to provide selective renal vasodilation in patients at risk for renal dysfunction. However, it is now clear that although low-dose dopamine may increase blood flow to the renal cortex, blood flow to the renal medulla may actually decrease.8 Given the relatively hypoxic environment of the renal medulla under normal circumstances (Chapter 1), this effect is potentially harmful. Furthermore, the increase in urine output that occurs with low-dose dopamine is due primarily to a direct tubular natriuretic effect rather than to renal vasodilation. In a well-conducted, large, randomized trial, low-dose dopamine did not reduce the incidence of acute renal failure in patients with early renal dysfunction.9 Dopamine has a number of other potentially detrimental effects, including inhibition of hypoxic ventilatory drive, impairment of ventilation-perfusion matching in the lung, and suppression of the secretion of some anterior pituitary hormones, such as prolactin, growth hormone, and thyrotropin.8
Dobutamine.
Dobutamine is useful for treating low cardiac output following cardiac surgery,10 and it may be used in combination with norepinephrine for treating septic shock.11 The dose range is 1 to 10 μg/kg/min. At higher doses tachyarrhythmias become common. Metabolic side effects are minimal.
Phenylephrine.
Phenylephrine is a noncatecholamine, direct-acting α receptor agonist that does not possess any significant β receptor activity. Bolus doses of 50 to 100 μg are commonly used during induction of anesthesia in patients with aortic stenosis to counteract the vasodilation produced by anesthetic drugs. In this situation phenylephrine has the advantage of increasing coronary perfusion pressure without increasing heart rate. Cardiac output may fall due to increased afterload and baroreceptor-mediated reflex bradycardia. Phenylephrine has a slightly longer duration of action than norepinephrine. The drug may be administered as a continuous infusion at a dose of 0.1 to 1 μg/kg/min.
Phosphodiesterase Type III Inhibitors
The phosphodiesterases (PDEs) are a family of enzymes that catalyze the breakdown of cyclic nucleotides, including cAMP and cGMP. There are multiple subtypes of PDE that have varying tissue distributions and actions.12 Caffeine and theophylline are nonspecific PDE inhibitors that are used as bronchodilators. Papaverine is a vasodilator and nonspecific PDE inhibitor that is used by cardiac surgeons during coronary artery bypass graft (CABG) surgery to prevent spasm of the internal mammary artery.
Drugs that selectively inhibit PDE subtype III function as inodilators. The commercially available PDE-III inhibitors all have a similar pharmacologic profile: they increase contractility and cause pulmonary and systemic (arteriolar and venous) vasodilation. As such, PDE-III inhibitors are useful for treating low cardiac output, particularly in the presence of pulmonary edema or pulmonary hypertension. They are potent vasodilators of coronary grafts13 and cause less tachycardia and atrial fibrillation than dobutamine.10 The inotropic effect is independent of the β1 receptor, which is advantageous in patients with β1 receptor desensitization (see earlier discussion). Also, by combining a PDE-III inhibitor with a β1 receptor agonist, a dual mechanism of action is exploited. Arteriolar and venous dilation can cause modest hypotension. This can be treated with either fluid or low-dose norepinephrine, depending on the status of the patient’s intravascular volume. In patients with cardiogenic shock, the combination of a PDE-III and norepinephrine provides support for both cardiac output and blood pressure, but without the troublesome tachycardia and metabolic disturbance that can occur with epinephrine.
PDE-III inhibitors are available as intravenous formulations for short-term use. Unlike the sympathomimetics, they have durations of action measured in hours, so their effects cannot be readily judged. Oral formulations of PDE-III inhibitors for the treatment of chronic heart failure have been studied but have resulted in higher mortality rates.14
Miscellaneous Vasoactive Drugs
Calcium.
An intravenous bolus dose of calcium chloride of 5 mg/kg (or 0.035 mmol/kg) increases blood pressure but has little effect on myocardial contractility.15 The duration of effect following a bolus dose is 5 to 10 minutes. The pressor effect is much more pronounced in the presence of hypocalcemia. Calcium chloride does not improve outcome after cardiac arrest16 and is no longer included in routine resuscitation protocols. However, calcium is useful in the management of hyperkalemia because it reduces potassium-induced arrhythmias, and in the treatment of hypocalcemia.
Vasopressin.
Vasopressin (V; antidiuretic hormone) is a peptide hormone released from the posterior pituitary in response to an increase in serum osmolarity or hypovolemia (Chapters 1 and Chapter 32). Stimulation of V1 receptors within vascular smooth muscle results in vasoconstriction (via the IP3/DAG second-messenger system), whereas stimulation of V2 receptors in the kidney results in water retention (via the cAMP second-messenger system). An additional action of vasopressin is to increase the release of the von Willebrand factor from the vascular endothelium, which increases platelet aggregation. The elimination half-time of vasopressin is 10 to 30 minutes.
Exogenously administered vasopressin is used to treat catecholamine-resistant vasodilatory shock. In early shock, endogenous stores of vasopressin are released from the posterior pituitary, but as shock progresses, these stores become depleted. In patients with advanced vasodilatory shock, vasopressin infusion at 4 units/hr combined with norepinephrine has been shown to be superior to norepinephrine alone in terms of hemodynamics and markers of splanchnic perfusion.17 However, higher doses of vasopressin, sufficient to replace rather than augment norepinephrine, can cause a marked reduction in cardiac output and may worsen splanchnic perfusion.18 In animal models, vasopressin is associated with relatively less vasoconstriction within the coronary, cerebral, and pulmonary circulations than is associated with catecholamines.19,20 Despite an antidiuretic effect, in patients with vasodilatory shock urine output may actually improve with vasopressin,21 presumably due to an improvement in renal blood flow. As with all vasoconstrictors, precipitous reductions in cardiac output can occur with vasopressin, particularly in the settings of hypovolemia and impaired ventricular function. Current recommendations are that vasopressin, in a dose of 0.01 to 0.04 units/min, should be considered in patients with vasodilatory shock who have adequate volume resuscitation and are refractory to high doses of catecholamines.22
Levosimendan.
Levosimendan is an inotropic drug that acts by stabilizing the interaction between calcium and tropo-nin, so-called calcium sensitization. In addition, levosimendan functions as a vasodilator, partly through PDE-III inhibition and partly by opening adenosine triphosphate gated potassium channels. Because the effect of levosimendan is not mediated by β1 receptors or increased cAMP, it does not increase myocardial oxygen consumption and is not arrhythmogenic. Levosimendan may be a particularly useful inotropic agent in patients who are receiving β blockers. In the treatment of heart failure, levosimendan is more effective than dobutamine in improving cardiac output and, in one study, was associated with better mortality compared to dobutamine.23
Levosimendan has an elimination half-time of 1 hour, but it has pharmacologically active metabolites with elimination half-times of up to 80 hours. The drug is usually administered as a continuous infusion of 0.1 to 0.2 μg/kg/min for 24 hours, which may be preceded by a loading dose of 12 to 24 μg/kg. Following a 24-hour infusion, a positive inotropic effect is sustained for several days. Side effects are typically modest and include mild tachycardia and hypotension. So far, levosimendan has been used primarily in patients with decompensated heart failure, but there is some limited experience in its use in patients after cardiac surgery.24
β Blockers
β Blockers tend to be either lipid-soluble (e.g., metoprolol) or water-soluble (e.g., atenolol). Lipid-soluble agents typically undergo extensive hepatic metabolism and have a low oral bioavailability. Some lipid-soluble β blockers are metabolized by the cytochrome P-450 (CYP) 2D6 enzyme system, so their metabolism is susceptible to inhibition by other drugs (see Table 4-3). In contrast, water-soluble agents have high oral bioavailability and tend to be eliminated unchanged by the kidney. In patients with hepatic impairment, a water-soluble β blocker is appropriate, whereas in patients with renal impairment, a lipid-soluble β blocker may be more appropriate.
β Blockers have antihypertensive, antiarrhythmic, and antiischemic actions, and they inhibit ventricular remodeling. Treatment with β blockers is associated with reduced mortality rates in patients with coronary artery disease25–28 and chronic heart failure (see Chapter 19) and in high-risk patients undergoing noncardiac surgery.29 In patients undergoing CABG surgery, preoperative treatment with β blockers is associated with reduced perioperative mortality rates.30 Acute cessation of chronic β blocker treatment can precipitate myocardial ischemia.31
In the cardiothoracic ICU, β blockers are used in the following circumstances:
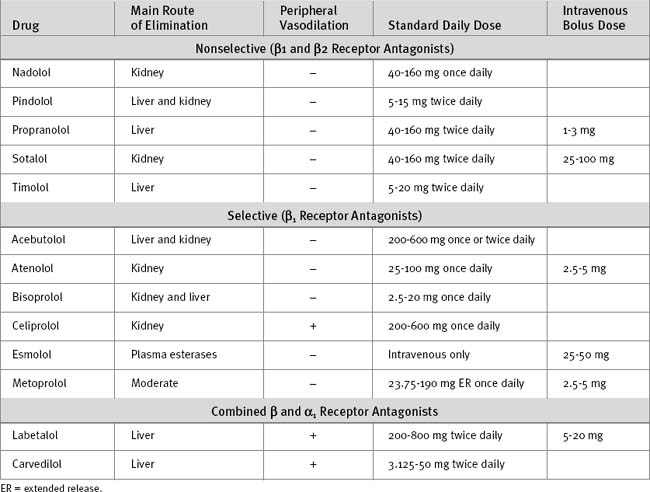
In patients with ventricular dysfunction, introduction of a β blocker may initially worsen the symptoms of heart failure. Ventricular remodeling and improved ejection fraction develop slowly over several months. Thus, β blockers should be avoided in patients who have only recently discontinued inotropic support or who are fluidoverloaded. Introduction of β blockers for the treatment of heart failure is generally not appropriate in the ICU. Characteristics of commonly used β blockers are listed in Table 3-2.
Labetalol.
Labetalol is an α1 blocker and a nonselective β blocker that is formulated for intravenous and oral use. Labetalol is a useful drug for the treatment of postoperative hypertension. Intravenously, labetalol may be administered as repeated bolus doses of 5 to 10 mg. Labetalol undergoes significant hepatic metabolism and has low oral bioavailability; thus, the oral dose is much higher than the intravenous dose.



