Cardiovascular Drug Interactions
Drug interactions occur when the combination of two or more medications alters the pharmacokinetic parameters or changes the pharmacologic response of either drug. These changes can produce undesirable responses including exaggerated or reduced pharmacologic effect or an added toxic response. It is clear that with the aging population and the increasing number of prescribed medications, the likelihood of having a significant drug interaction increases. In general, drug interactions account for a reported 7% to 17% of all adverse drug events and are probably higher considering underreported events.
Drug interactions are categorized as either pharmacokinetic or pharmacodynamic. Pharmacokinetic interactions occur when combining two or more medications results in an alteration of the drug’s disposition in the body. Examples include the use of amiodarone with warfarin resulting in an increased international normalizing ratio (INR) secondary to decreased hepatic metabolism of warfarin, or itraconazole decreasing the metabolism of certain HMG-CoA reductase inhibitors. Pharmacodynamic interactions occur when the addition of a medication leads to changes in the pharmacologic response of either medication. Examples include the addition of digoxin for heart rate control to beta-blockers or nondihydropyridine calcium channel blockers, which may lead to an unacceptable lowering of heart rate.
This chapter outlines typical types of drug–drug interactions observed in clinical practice and includes information on selected food–drug interactions that are commonly encountered.
PHARMACOKINETIC DRUG INTERACTIONS
As reviewed in Chapter 60, the basic characteristics of pharmacokinetics include absorption, distribution, metabolism, and elimination. Alterations in one or more of these characteristics may lead to a significant drug interaction. Many of the documented interactions occur as a result of changes in metabolism or elimination of medications.
Absorption-related interactions result in decreases or increases in the amount of drug absorbed as well as delays in absorption. The presence of food or certain types of food may change medication absorption characteristics, leading to either a decrease in the extent or an increase in absorption time. Certain medications must be taken on an empty stomach for adequate bioavailability. For example, captopril, a non–prodrug angiotensin-converting enzyme inhibitor, is a prototypical drug that must be taken on an empty stomach because food may decrease the bioavailability by 25% to 50%. Additional medications that should be taken on an empty stomach are listed in Table 62.1. In addition, other medications such as bile acid binders and fiber laxatives may alter absorption of medications. Bile acid binders such as cholestyramine interfere with the absorption of a number of medications, and as a general rule, medications should be taken 2 hours before or 2 hours after the bile acid resin to minimize a decrease in absorption. Another mechanism that may alter absorption is chelation, whereby di- or trivalent cations such as calcium and aluminum bind and decrease bioavailability. Tetracycline and quinolone antibiotics are prototypical for chelating interactions. Also, medications that may increase gastric motility, such as metoclopramide and erythromycin, may alter the bioavailability of medications because gastrointestinal transit times are hastened.
TABLE
62.1 Cardiovascular Medications that should be Taken on an Empty Stomach

Certain medications require either an acidic or basic gastrointestinal pH for absorption. Alterations in pH may change the bioavailability of these medications. For example, itraconazole requires an acidic pH for optimal absorption, and medications such as H2-blockers and proton pump inhibitors may decrease bioavailability and possibly result in treatment failure. Finally, antibiotics can alter the flora of the gastrointestinal tract and may change the bioavailability or efficacy of certain medications. Digoxin is metabolized in the gastrointestinal tract by the bacteria Eubacterium lentum in approximately 10% of patients. Both tetracycline and erythromycin can decrease the levels of this bacterium, therefore increasing the bioavailability of digoxin. Antibiotic-induced vitamin K depletion may interfere with warfarin. Gut flora that produce vitamin K may be killed, leading to an increased sensitivity to the effects of warfarin.
Alterations in protein binding may also play a role in drug–drug interactions. Drugs that are highly protein or tissue bound are more affected by other drugs that displace the original drug from the protein-binding site. Levels of the unbound fraction of drug will increase either transiently or permanently when the two medications are given concomitantly. These interactions are more difficult to identify, and medications with high protein binding such as warfarin and digoxin should be taken into consideration when adding or discontinuing medications.
Drug metabolism occurs via phase I or II reactions. Phase I reactions include oxidation, reduction, or hydrolysis and convert parent compounds into more water-soluble compounds such as losartan. These metabolites may be inactive, have more activity than the parent compounds (prodrugs), or have less activity than the parent compound. Phase II reactions typically result in development of inactive compounds via glucuronidation, sulfation, or addition of other endogenous substances such as lorazepam.
Most phase I reactions occur via the cytochrome P450 (CYP) enzymes found in the liver, gastrointestinal tract, brain, kidneys, and other organs throughout the body. The vast majority of the enzymes are hepatic; however, there are large concentrations in the gastrointestinal tract as well. The CYP enzymes are a group of heme-containing compounds located on the membrane of the endoplasmic reticulum. The nomenclature for CYP includes a lead number referring to the family, followed by a letter referring to the subfamily, and finally an additional number that refers to the individual enzyme. Examples include CYP3A4 or 2D6. Most drug metabolism occurs via enzymes in the families 1, 2, and 3. CYP3 enzymes account for nearly 70% of the total CYP in the liver.
An individual drug can be a substrate, inhibitor, or inducer for a specific enzyme. A drug may act as an inhibitor of one or more groups of enzymes and may be a substrate for one or more groups of enzymes. For example, amiodarone is a substrate for CYP3A4 and is an inhibitor of CYP2C9 and 2D6 enzymes. Genetic variation also exists in the expression of certain CYP enzymes. Polymorphism is seen with both CYP2D6 and 2C19, and expression of the enzyme can be variable. Between 3% and 10% of Caucasians and 2% of Asians and African Americans have either low or no activity of CYP2D6. Patients with low or no activity of a particular isoenzyme are known as “poor metabolizers.” Some individuals are also poor metabolizers of medications that are eliminated through the CYP2C19. Many commonly used cardiovascular medications are eliminated through the CYP system. Table 62.2 lists the drugs and the enzyme(s) responsible for their elimination as well as other enzyme(s) they may inhibit.
TABLE
62.2 Commonly Observed Drug–Drug Interactions with Cardiovascular Medicationsa
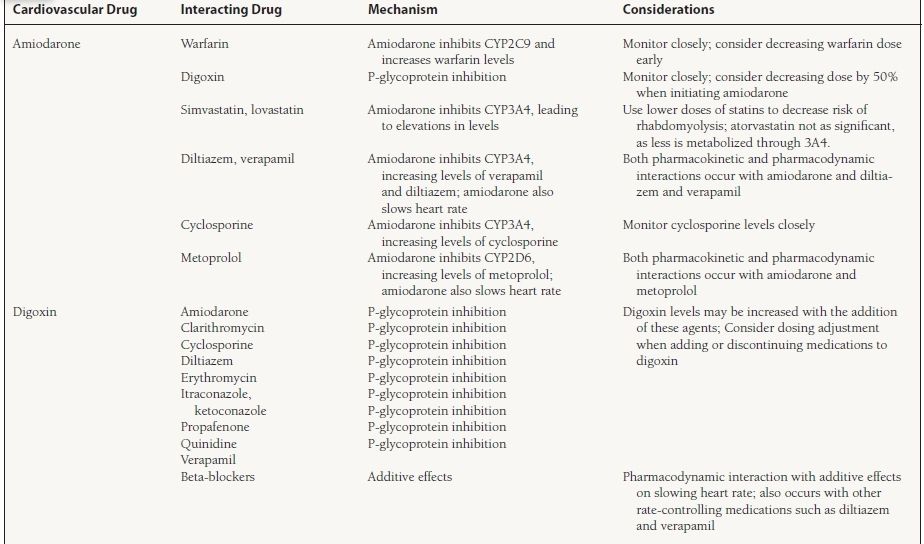

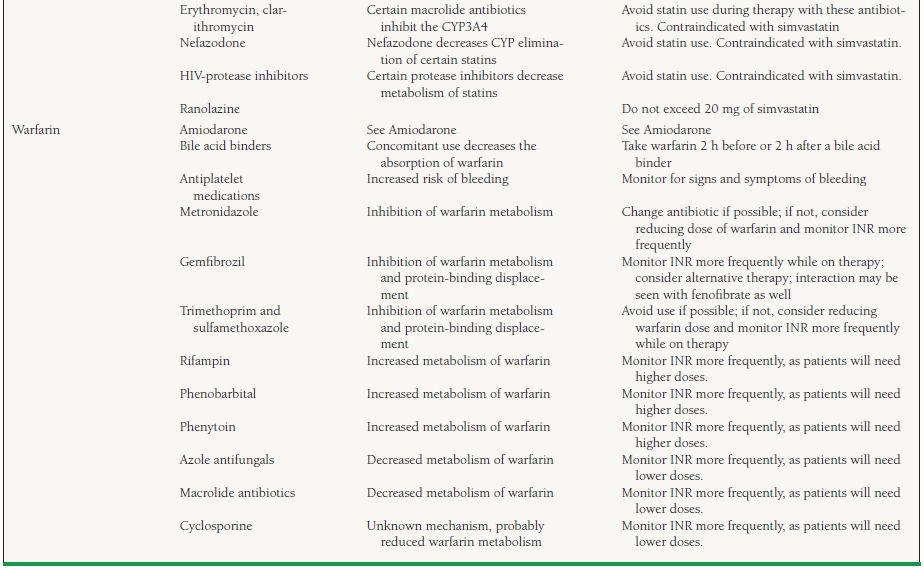
a This table provides a limited list of medications and interactions and is by no means complete. A review of the complete medication list for drug interactions should be performed frequently, especially when adding or discontinuing medications, to avoid unnecessary adverse drug events.



