Fig. 2.1
Main inflammatory mechanisms participating in atherosclerotic lesion initiation, progression, and plaque destabilization and rupture
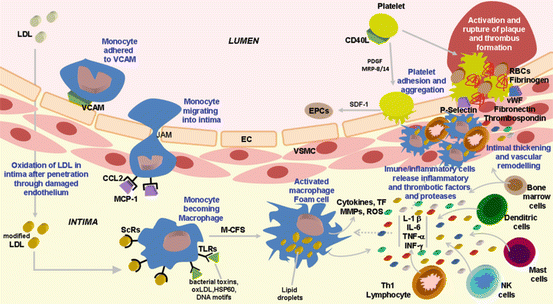
Fig. 2.2
Vascular and immune inflammatory cells that participate in atherosclerosis and main molecular mediators involved in atherosclerotic lesion initiation, progression, and plaque destabilization and rupture
It has been suggested that the early phase of atherosclerosis is better explained as an inflammatory response caused by retention of lipoproteins in the arterial intima. The increased amount of cholesterol-rich very low-density lipoprotein (VLDL) and low-density lipoprotein (LDL) in circulation, and the existence of a damaged endothelium, elicits lipoprotein infiltration in the artery wall and accumulation in the extracellular matrix. This accumulation exceeds the capacity for elimination, and this is the initiating pathological process of atherosclerosis [2]. LDL oxidation in the intima causes release of bioactive phospholipids that activate endothelial cells (ECs), which then express several types of leukocyte adhesion molecules, such as the vascular cell adhesion molecule-1 (VCAM-1), to which monocytes and lymphocytes will preferentially adhere, at sites of hemodynamic strain [15]. This vascular inflammation is propagated by chemokines, which are inflammatory cytokines responsible for the recruitment of distinct leukocyte classes to the atheroma; a variety of different chemokines mediate chemotactic recruitment of adherent monocytes and lymphocytes to the forming lesion [3, 16]. In human atherosclerotic lesions, enhanced expression of several chemokines was found, including CXC chemokines (such as IL-8 [IL-8/CXCL8], neutrophil-activating peptide-2 [NAP-2/CXCL7], growth-related oncogene-α [GRO-α/CXCL1], interferon [INF]-γ-inducible10 [IP-10/CXCL10], and CXCL16) and CC chemokines (such as monocyte chemoattractant protein-1 [MCP-1/CCL2], leukotactin-1 [Lkn-1/CCL15], regulated on activation, normal T cell expressed and secreted [RANTES/CCL5], CCL19, and CCL21); fractalkine (CX3CL1), a membrane-bound chemokine, is also expressed and promotes recruitment of CX3CR1+ mononuclear cells to the lesion area.
Transmigration of monocytes occurs mainly in areas where the basal lamina is enriched with modified LDL and via endothelial cell junctions (junction adhesion molecule [JAM]-A and [JAM]-C), which are implicated in the control of vascular permeability and leukocyte transmigration (Fig. 2.2) [17]. Chemokines have been viewed as the link between lipidic and inflammatory pathways in atherogenesis, which is supported by the fact that modified LDL particles elicit chemokine stimulation in macrophages and vascular smooth muscle cells (VSMCs), among other cells [3, 16].
Chemoattractant factors, which include monocyte chemoattractant protein-1 (MCP-1 or CCL2) produced by vascular wall cells in response to modified lipoproteins, are responsible for the migration and diapedesis of adherent monocytes [16]. Monocytic cells directly interacting with human ECs increase severalfold the production of monocyte matrix metalloproteinase 9 (MMP-9), leading to subsequent infiltration of leukocytes through the endothelial layer [18]. The inflamed intima produces the macrophage colony-stimulating factor (M-CSF), a cytokine/growth factor that is responsible for the differentiation of monocytes into macrophages [15]. This process is associated with macrophage overexpression of pattern recognition receptors for innate immunity, including scavenger receptors (ScRs) and Toll-like receptors (TLRs) (Fig. 2.2) [19]. ScRs use receptor-mediated endocytosis to engulf and degrade a broad range of molecules and particles carrying pathogen-like molecular patterns, including oxidized lipoproteins (in particular, oxidized LDL [oxLDL]), bacterial endotoxins, and apoptotic cell fragments. Accumulation of cholesterol esters in the cytoplasm droplets, aggravated by pro-inflammatory cytokines and endotoxins that inhibit the expression of ATP-binding cassette transporters ABCA1 and ABCG1 responsible for cholesterol efflux from the cell, converts macrophages into foam cells, which are lipid-laden macrophages typical of early-stage atherosclerosis. On the other hand, and unlike ScRs, TLRs do not mediate endocytosis, but bind ligands with pathogen-like molecular patterns (such as bacterial toxins, oxLDL, stress proteins such as heat shock protein 60 [HSP60], and DNA motifs), initiating a cascade that culminates in macrophage activation [19]. This process also occurs in dendritic cells, mast cells, and endothelial cells, all of them possessing TLRs (Fig. 2.2).
Additional mechanisms contribute to the inflammatory milieu within the lesion that determines plaque formation, including advanced glycation end products (AGE) that, after binding to receptors for AGE (RAGE) on macrophages, ECs, and VSMCs, causes amplification of vascular inflammation due to activation of nuclear factor kappa B (NF-κB). Moreover, activated macrophage produces reactive oxygen species (ROS) and triggers the release of vasoactive molecules, including endothelins, eicosanoids, and nitric oxide, altogether contributing to amplify lipoprotein oxidation and cytotoxicity [15]. These macrophages also secrete proteolytic enzymes that are able to degrade matrix components, thus having a later role on destabilization of plaques and contributing to an increased risk for plaque rupture and thrombosis (Figs. 2.1 and 2.2).
Role of Inflammatory Mediators in Atherosclerotic Lesion Progression
Whereas foam cell accumulation characterizes fatty streaks, deposition of fibrous tissue defines the more advanced atherosclerotic lesion [20]. Macrophage-derived foam cells are crucial in lesion progression because they amplify the inflammatory response through the secretion of numerous growth factors and cytokines, including tumor necrosis factor-α (TNF-α) and interleukin (IL)-1β (Fig. 2.2). In addition, T lymphocytes join macrophages in the intima and govern the adaptive immune responses. These cells, as well as ECs, secrete additional cytokines and growth factors that promote the migration and proliferation of VSMCs (Fig. 2.2). In response to inflammatory stimulation, VSMCs express specialized enzymes that can degrade elastin and collagen, allowing their penetration into the expanding lesion.
Chemokines within the plaque elicit activation of ECs and different leukocyte subsets (such as T cells), causing additional release of inflammatory cytokines and chemokines, which then promote further recruitment and activation of leukocytes, in a cycle of amplification of inflammation [3, 16]. Chemokine receptor CXCR3 interacts with its ligands (CXCL9 [MIG], CXCL10 [IP-10], and CXCL11 [I-TAC]) and promotes an inflammatory T-cell phenotype that causes macrophage activation [16]. Activation of macrophages also occurs directly by chemokines, which trigger increased release of cytokines and production of tissue factor (TF), MMPs, and ROS, and enhanced lipid loading and foam cell formation by upregulation of scavenger receptors. Altogether, these mechanisms, and the interaction between MCP-1 and CCR2, will highly alter macrophages to a pro-inflammatory, matrix-degrading, procoagulant, and proapoptotic phenotype [16].
Beyond their effects on T cells and macrophages, chemokines also interfere with VSMCs. While chemokines may promote VSMC migration into the lesion, an early event in atherogenesis, they could also transform these cells from a nonproliferative contractile phenotype, typical in healthy arteries, into actively proliferative cells (synthetic phenotype), which migrate when attracted by chemotactic agents, and increase matrix synthesis [16]. The migration of VSMCs from the vascular media to the intima is a hallmark process in intimal thickening and vascular remodeling characterizing atherosclerosis (Fig. 2.1). Recent data suggests that circulating bone marrow cells and progenitor cells present in the adventitia may also be a potential source of VSMCs in the intima [21] (Fig. 2.2).
T lymphocytes have been shown to contribute to atherogenesis and lesion development and progression [22, 23]. T cells are recruited to the forming lesion by mechanisms similar to those described for monocytes, despite using different patterns of activation, which is dependent upon recognition of cognate antigens and concomitant ligation of costimulatory receptors. Adhesion molecules (such as VCAM-1) and chemokines trigger T cells to enter the atheroma, where they react to several local molecules (peptide antigens) bound to major histocompatibility complex (MHC) molecules on the surface of antigen-presenting cells (APCs), such as oxLDL, HSP60, and microbial antigens. T-cell activation elicits distinct responses, involving T-helper-1 (Th1) cells, regulatory T cells (Treg), Th2 cells, and natural killer (NK) T cells [15, 22]. The precise role played by Th2 cells remains to be elucidated, while NK T cells recognize lipid antigens presented by CD1 molecules and trigger responses identical to those elicited by MHC-restricted T cells, including release of pro-inflammatory mediators and progression of lesion. While Th1 response, the most common in atheroma, involves induction of pro-inflammatory cytokines and promotion of lesion formation, Treg cells have an inhibitory effect on this process. In fact, Th1 cells produce IFN-γ, TNF-α, and CD40 ligand (CD40L), among other mediators, which are responsible for several responses. IFN-γ elicits activation of macrophages and endothelial cells via production of adhesion molecules, cytokines, chemokines, radicals, proteases, and coagulation factors, thus contributing to atherogenic lesion progression. TNF-α is produced by Th1 cells, but also by macrophages and NK cells, and has pro-inflammatory effects via NF-κB pathways [15, 22]. Other cytokines also play important roles in the atherogenic process [3, 16]. IL-18 receptor is expressed in endothelial and VSMCs, as well as in macrophages, and this cytokine elicits several important actions in atherogenesis, including induction of adhesion molecules (namely, VCAM-1), chemokines (i.e., IL-8), cytokines (in particular, IL-6), and several [1, 9, 13] MPPs, and, in combination with IL-12, upregulates the expression of IFN-γ in T cells, in macrophages, and in VSMCs, thus influencing important pro-inflammatory pathways involved in atherogenesis [16]. In opposition to Th1 cells, Treg cells are implicated in the maintenance of self-tolerance and regulation of autoimmunity, and the anti-inflammatory properties seem to derive from the production of transforming growth factor beta (TGF-β) and/or IL-10. T cells in atheroma display an activated/memory phenotype, and the proportion of activated T cells is particularly high in advanced lesions causing acute coronary syndrome (ACS).
The contribution of vascular dendritic cells (DCs) and mast cells to atherogenesis progression has been also suggested [24]. Although the exact role of vascular DCs remains poorly understood, they are thought to show antigen to naïve T cells accelerating lymphocyte recruitment into the atherosclerotic vessel. Mast cells seem to coordinate both the innate and acquired immunity through the activation of TLRs and cytokine release (Fig. 2.2).
Platelets also play a major role in the pathogenesis of atherosclerosis by several mechanisms, including production of inflammatory mediators (such as CD40L, myeloid-related protein-8/14 [MRP-8/14], and platelet-derived growth factor [PDGF]), and elicit leukocyte adhesion and incorporation into plaques, which is a robust example of the interplay between inflammation and thrombosis in the biology of atherothrombosis (Fig. 2.2) [20]. Platelets, as well as other cells involved in the disease (ECs and VSMCs, macrophages, and T cells), express CD40L and its receptor CD40 [25]. This pro-inflammatory cytokine plays a relevant role in this stage of atherogenesis, including stimulation of expression of adhesion molecules and of secretion of cytokines and MMPs which are involved in the degradation of extracellular matrix, as well as expression of tissue factor in ECs, VSMCs, and macrophages, which is responsible for the initiation of the coagulation cascade when exposed to factor VII. Thus, CD40L participates in the molecular events that will contribute to destabilization of plaques and increases the risk for plaque rupture and thrombosis.
Inflammatory Activation and Rupture of Plaque
Plaque rupture and the ensuing thrombosis commonly cause the most serious acute complications of atherosclerosis. Recent data suggest that inflammatory mechanisms, including infiltration and activation of leukocytes into the atherosclerotic plaque, rather than the degree of stenosis, are involved in plaque destabilization and rupture, resulting in coronary thrombosis leading to myocardial ischemia and infarction, for example (Fig. 2.1). In fact, ACS patients present typical features of an inflammatory phenotype, presenting several blood disturbances including increased contents of acute-phase reactants, cytokines, and activated T cells.
Plaque destabilization seems to occur due to the action of several types of molecules, including pro-inflammatory cytokines, chemokines, proteases, coagulation factors, vasoactive molecules, and radicals, mainly produced by activated macrophages, T cells, and mast cells located at sites of rupture (Fig. 2.2) [15, 16, 20]. These mediators are involved in several mechanisms that promote plaque activation, rupture, thrombosis, and ischemia, including inhibition of stable fibrous cap formation, degradation of collagen cap, and initiation of thrombus formation. Chemokines have been associated with the immune-mediated plaque destabilization due to several distinct mechanisms, namely, recruiting of activated leukocytes, stimulation of matrix degradation by macrophages, and induction of tissue factor in vascular ECs and VSMCs, as well as by promotion of oxidative stress and apoptosis in the plaques [16]. In addition, besides their role in thrombus formation, platelets could also have a major role on ACS, namely, due to release of pro-inflammatory mediators, including chemokines (such as CCL5, CXCL1, CXCL7, and epithelial neutrophil-activating peptide-78 [ENA-78/CXCL5]), and due to exacerbated inflammation in leukocytes and ECs [16].
The inflammatory milieu within atherosclerotic plaques stimulates the degradation of collagen fibers and inhibits the formation of new collagen, thus impairing the integrity of the interstitial collagen of the fibrous cap. In addition, activated T cells secrete IFN-γ which inhibits production of collagen by VSMCs. T lymphocytes are involved in several mechanisms that contribute to plaque destabilization, including production of IL-1 and CD40L which elicits macrophages to release interstitial collagenases, including MMPs [1, 8, 9, 13], that were detected in the shoulder region of plaques as well as in areas of foam cell accumulation, where activation and rupture of the plaque occur [22, 23]. MMPs are involved in the deregulation of extracellular matrix and intravascular thrombus formation, due to overexpression of tissue factor and activation of the coagulation cascade, thus contributing to plaque rupture [26].
Proteolytic destruction of collagen and inhibition of VSMC growth cause the reduction of cap thickness and strength, which become unable to support the hemodynamic forces and fissures, thus exposing components of the vascular matrix, including, e.g., different types of collagen, von Willebrand factor (vWF), fibronectin, laminin, fibulin, and thrombospondin (Fig. 2.2) [24]. ACS widely occurs due to physical disruption of the fibrous cap, either frank cap fracture or superficial endothelial erosion. The contact of blood with thrombogenic material (platelets and coagulation factors) initiates the formation of a thrombus, which can lead to abrupt and impressive obstruction of blood flow through the affected artery, and ischemia occurs in the end organ, such as the myocardium or the brain [20]. When the thrombus is nonocclusive or transient, the result could be clinically silent or cause symptoms typical of an ACS, including acute MI, unstable angina, or sudden death. The concentrations of circulating plasminogen activator inhibitor 1 (PAI-1) and fibrinogen usually determine the fate of a given plaque disruption [20].
Inflammatory Mediators as Biomarkers in Cardiovascular Disease
Considering the role played by inflammation in all the stages of atherosclerotic disease, a variety of inflammatory mediators have been tested, in experimental and in clinical studies, as putative biomarkers of the pathogenic mechanisms that underlie the initiation and evolution of the disease, as well as predictors of risk and future events. The main question is whether plasma markers of inflammation can be noninvasively and feasibly used to improve diagnosis and prognosis of distinct forms of atherosclerosis.
The putative biomarkers of inflammation include different types of molecules, such as acute-phase reactants (namely, C-reactive protein [CRP], serum amyloid A (SAA) proteins, and fibrinogen), cytokines (including IL-1β, TNF-α, IL-6, and IFN-γ), adipokines (i.e., adiponectin), adhesion molecules (such as selectins and VCAM-1), platelet products (namely, CD40 ligand and MRP-8/14), and proteases (namely, MMP-9) [15, 16, 20]. Some of these analytes may participate in a pathogenic pathway, but not acting as an effective biomarker, while others may reflect the local inflammatory process in the artery, but do not have a causal role on the disease (thus acting as a biomarker, but not as a risk factor).
CRP has been viewed as the prototype of inflammatory markers. Accumulated evidence suggests that it could be a feasible marker of cardiovascular risk in MI patients, which presents augmented levels, and a predictor of future events in apparently healthy individuals [27, 28]. The development of a high-sensitivity assay for CRP (hsCRP) allowed accurate measurement of this molecule as a tool to enhance risk stratification. In fact, hsCRP has been shown predictive value beyond that of traditional risk factors, at least in specific populations with intermediate risk according to conventional criteria (namely, the Framingham algorithm), to whom the determination of hsCRP was already suggested (by US Centers for Disease Control and Prevention [CDC] and American Heart Association [AHA]) as a way to improve identification and stratification of risk [29]. Note that this intermediate risk group accounts for much of the burden of cardiovascular events recommending improved prediction tools. The Reynolds risk score adds hsCRP (as well as the family history of premature CAD) to traditional risk factors in a clinically useful manner [30].
Considering a successful application to identify and stratify cardiovascular risk, as well as its independence from traditional risk factors (namely, hypercholesterolemia), biomarkers of inflammation were suggested as a way to identify individuals who might benefit from therapeutic intervention, regardless of low estimates of cardiovascular risk according on traditional risk factors. The JUPITER (Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin) trial enrolled more than 17,000 individuals without known CVD, low LDL-c levels (<130 mg/dL), and hsCRP >2 mg/L in order to evaluate the effect of statin therapy in cardiovascular events and mortality [31, 32]. The results showed a major (>40 %) reduction of events and all-cause mortality, and additional analyses suggested that the benefits could be derived both from LDL reduction and improved anti-inflammatory effect viewed by hsCRP reduction. Evidence that a direct anti-inflammatory therapy positively impacts on atherosclerotic events cannot be drawn from the study, but ongoing trials (Canakinumab Anti-Inflammatory Thrombosis Outcomes Study [CANTOS] and Cardiovascular Inflammation Reduction Trial [CIRT]) could provide evidence on this, which is expected to have a major impact in the way CVD could be managed in the near future.
All these putative useful roles played by hsCRP need to be precisely defined through adequate and larger clinical trials that may test a number of targets and mediators. At the moment, doubts remain whether CRP could alone reflect the inflammatory mechanisms underlying atherosclerosis development, especially knowing the involvement of several other mediators (including a variety of cytokines), whether CRP could be viewed as a risk factor (with a causal role) or just as a surrogate marker of the process, and finally whether CRP or any other emergent biomarker of inflammation and cardiovascular risk could effectively and consistently improve clinical performance (namely, diagnosis, prediction of risk and/or of mortality, therapeutic decisions) in distinct populations.
The validation of the concept that inflammation is crucial for atherogenesis and its translation to clinical practice is an ongoing mission that deserves continuous efforts.
Inflammatory Mechanisms Involved in Atrial Fibrillation
Pathophysiology of Atrial Fibrillation
The development of abnormal tissue substrate allows multiple reentrant wavelets of excitation to propagate within the atrial myocardium contributing for the maintenance of AF [33]. As AF episodes get more frequent and/or prolonged, restoring and maintaining sinus rhythm becomes more difficult – “AF begets AF” [34]. This is the result of electrical remodeling and electrophysiological changes such as shortening of the atrial effective refractory period and action potential duration [33]. During structural remodeling, myocyte degeneration and myocardial fibrosis occur, leading to left atrial enlargement [34]. These changes create a susceptible substrate that has been implicated in the perpetuation of AF.
Garrey in 1914 proposed the “critical mass theory” of fibrillation. This hypothesis went nearly unchallenged for over 60 years and was based on the fact that multiple, changing reentrant circuits, modified by refractory tissue areas, caused the irregular activity seen during AF [35]. According to this theory, there is a critical mass above which AF is sustained, meaning that the greater the atrial tissue surface area, the higher the probability of having sustained episodes of arrhythmia [35]. This mass is correlated with the area of geometric/structural and functional abnormalities. However, almost 20 years ago, Haïssaguere and colleagues in a seminal investigation identified pulmonary veins as the initiators of AF episodes and first assessed radiofrequency ablation as an effective way of treating this arrhythmia [36].
Inflammation is associated with electrophysiological and structural atrial remodeling and facilitates the disease development and perpetuation [33]. Several histological findings confirming the presence of active inflammation in patients with AF are known to occur. Nakamura et al. found active atrial perimyocarditis with inflammatory infiltrates, lipid degeneration, and fibrosis in patients with nonvalvular AF and cardiogenic thromboembolism [37]. Frustaci et al. have shown in atrial biopsies from patients with AF a higher prevalence of inflammatory infiltrates, myocyte necrosis, and fibrosis, whereas biopsies from control subjects were normal [38]. More recently, Narducci et al. assessed atrial inflammation by the identification of CRP in atrial cardiomyocytes. CRP was more frequent in paroxysmal rather than in persistent AF patients and wasn’t present in the control group [39].
The possible association between AF and inflammation is suggested by several studies based on the identification of inflammatory serum biomarkers that are elevated in patients with AF. Considering the existent published data, inflammation may be linked with the initiation and perpetuation of AF and also with the increased thrombotic burden of this population [40].
Inflammatory Markers in Atrial Fibrillation
Table 2.1 presents the main studies involving inflammatory markers and atrial fibrillation.
Table 2.1
Main studies involving inflammatory markers and atrial fibrillation
Author/reference | Population and study design | Results |
|---|---|---|
Aviles et al. (2003) [41] | Epidemiologic, cross-sectional study of 5,806 subjects, longitudinal study of 5,491 | CRP associated with the presence of AF; CRP predicts patients at ↑ risk for future development of AF |
Chung et al. (2001) [42] | Retrospective, observational case–control study, 131 patients with atrial arrhythmia and 71 controls | ↑ CRP in AF, CRP ↑greater in persistent AF than paroxysmal AF |
Dernellis and Panaretou (2004) [43] | Prospective case–control study, 50 patients with AF and 50 controls | ↑ CRP in AF, CRP inversely related to successful cardioversion rate |
Watanabe et al. (2005) [44] | Prospective, 50 AF patients | ↑ CRP in PAF vs. control group; L-PAF had higher CRP levels than S-PAF; both S-PAF and L-PAF had larger LA diameter than control subjects; L-PAF > S-PAF in LA size |
Asselbergs et al. (2005) [45] | Cross-sectional study, 8,501 patients | CRP and microalbuminuria are independent risk factors for AF; 2) both factors together represent a fourfold higher risk |
Acevedo et al. (2003) [46] | Prospective, 109 patients with AF | ↑ CRP in AF patients compared with control group at baseline; during follow-up, CRP in patients still in AF was significantly ↑ when compared with patients in SR |
Psychari et al. (2005) [47] | Prospective, 90 patients with persistent and permanent AF and 46 controls | IL-6 and CRP participate in the evolution of AF; 2) LA size correlates with CRP and IL-6 levels |
Conway et al. (2003) [39] | Cross-sectional observation study, 106 AF patients and 41 controls | ↑ IL-6, CRP, and plasma viscosity, correlating to a prothrombotic state; ↑ IL-6 and CRP were independent predictors of stroke and composite end point of stroke or death |
C-Reactive Protein and the Development of Atrial Fibrillation
CRP is a marker of the magnitude of inflammation and is widely used in the clinical setting [41]. It has been used to detect acute injury, infection, and inflammation [41, 48]. It is an acute-phase protein produced in the liver in response to IL-1, IL-6, and TNF-α that binds to damaged cells, activating the complement pathway [41].
CRP increase correlates with higher risk of cardiovascular events [49]. The exact mechanism beneath the association of CRP with AF is uncertain. This protein may be responsible for starting or perpetuating AF or can possibly just be a marker of underlying disease [42]. Figure 2.3 schematically summarizes the impact of elevated CRP levels in atrial fibrillation patients. It specifically binds to phosphatidylcholine on the membranes of myocardial cells activating the complement system and, in the presence of Ca2+ ions, leads to the formation of new molecular intermediaries responsible for cellular membrane dysfunction through changes in transmembrane ion transport [42]. CRP can also induce apoptotic loss of atrial myocytes due to calcium accumulation within atrial myocytes and may also play a role in the clearance of apoptotic atrial myocytes as an opsonin [42]. Since myocyte loss is typically accompanied by replacement fibrosis, inflammation provides substrate for AF development [42].
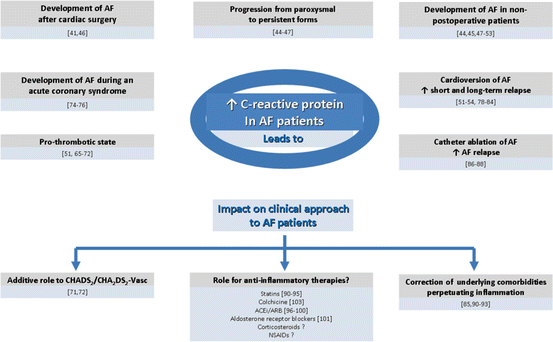
Fig. 2.3
Impact of elevated hsCRP levels in atrial fibrillation patients
Serum CRP levels are significantly higher in patients with paroxysmal and chronic AF than in normal controls [50]. Similarly, CRP levels are higher in chronic AF patients than in paroxysmal AF patients [50]. The first investigation linking elevated CRP levels and AF was reported by Bruins and colleagues in the setting of cardiac surgery. They verified that patients who developed AF, mostly on the second/third postoperative day, concomitantly presented a peak elevation of CRP [51]. Years later in a small retrospective study, Aviles et al. demonstrated that patients who developed AF after major thoracic surgery had a nearly twofold increase in postoperative CRP levels in comparison with control subjects [41].
The relation between AF and CRP in non-postoperative patients was firstly reported by Chung and colleagues in a case–control study [42, 52]. CRP levels were more than twofold higher in patients with AF compared with controls. Patients with permanent AF also had higher levels of CRP compared with paroxysmal AF patients suggesting that the role of inflammation in AF may be more relevant in promoting persistence rather than initiating AF [42, 53].
Most of the previously reported studies consider patients with a moderate to high cardiovascular risk, with other coexisting cardiovascular diseases that may be the cause for elevated inflammatory markers [54]. In the Women’s Health Study, all of the participating 24,734 women were free from cardiovascular disease and AF at baseline. In this study, inflammation measured through the levels of CRP, ICAM-1 (intercellular adhesion molecule 1), and fibrinogen was significantly associated with the risk of incident AF [54].
Acevedo et al. conducted a study with 130 patients with newly diagnosed nonvalvular AF [46]. In this population, CRP levels were predictors of AF at 30 days and at 1-year follow-up. Patients who reverted to sinus rhythm had lower baseline CRP levels. These low levels were kept during the first year of follow-up [46]. Patients who stayed in AF had higher baseline and 1-year follow-up CRP levels [46].
Psychari et al. correlated elevated CRP with AF and with left atrial size, suggesting a role for inflammation in AF as being a part of the atrial remodeling process and hence an important trigger [47]. Increased left atrial size and dysfunction were also correlated with higher CRP levels [44]. Watanabe et al. suggested that longer AF duration was associated with CRP elevation and an increased left atrial appendage diameter [44].
The association between AF and autoimmune diseases like Graves’ and celiac disease and psoriasis suggests that AF could be a T-cell autoimmune disorder involving cytotoxic and helper T cells acting on His-Purkinje cells, which express contactin-2 [55]. Interestingly, this is one of the mechanisms known to be behind multiple sclerosis, and patients with this disease also have a higher incidence of atrial fibrillation [55]. However, since most of the His-Purkinje cells are in the ventricles, this relation is doubtful [55].
Interleukins and AF
The interleukin (IL) system is the main modulator of the inflammatory response [56]. IL-1β and IL-1Ra are the two major cytokines regulating inflammatory response. Genetic polymorphisms of IL-1 cluster genes are associated with increased risk of inflammatory and cardiovascular disease [56, 57]. VNTR (variable number tandem repeat) polymorphisms of the IL-1RN gene were associated with lone AF and also with several other chronic inflammatory diseases like ulcerative colitis, periodontitis, osteoarthritis, and gastric cancer [56, 57].
IL-6 is a cytokine involved in the synthesis of acute-phase proteins such as CRP. High plasma IL-6 levels have been correlated with the presence and duration of AF and increased left atrial diameter [58]. Both higher levels of IL-6 and several genetic polymorphisms, namely, in the promoter region of the IL-6 gene, were related with AF genesis in postoperative patients. Patients with AF and coronary disease had more frequently the CC genotype of 174G/C IL-6 polymorphisms. IL-8 (a cytokine that acts as a powerful chemoattractant for neutrophils) levels were also found to be elevated in AF patients [58].
Transforming Growth Factor
Transforming growth factor beta-1 (TGF-β1) is an important molecule promoting fibrosis in the liver, lung, kidney, pancreas, and other organs [59, 60]. It has been shown that it may also play an important role in the process of cardiac fibrosis, inflammation, and tissue remodeling organs [59, 60]. The TGF-β1 pathway is activated in AF; it can promote atrial fibrosis, inflammation, and tissue remodeling and then lead to tissue fibrosis [61].
Pentraxin 3
Pentraxin 3 is produced in local inflammatory lesions by various cells, namely, monocytes and macrophages, endothelial cells, vascular smooth muscle cells, fibroblasts, dendritic cells, and adipocytes [62]. Soeki et al. demonstrated that, in AF patients, the production of pentraxin 3 in the left atrium might reflect local inflammation; also in the GISSI-AF trial (Use of Valsartan, an Angiotensin II AT1-Receptor Blocker in the Prevention of Atrial Fibrillation Recurrence), higher concentrations of pentraxin 3 were detected in patients with AF, compared with those in sinus rhythm [62].
Oxidative Markers and Atrial Fibrillation
Emerging evidence implicates oxidative stress in the initiation and maintenance of AF. Oxidative stress promotes calcium overload and myocardial stunning alongside with decreased nitric oxide (NO) bioavailability [63]. Atrial redox balance results in a significant reduction in contractile response to β-adrenergic stimulation [63].
In experimental models, Carnes et al. have shown that AF induced in dogs decreases tissue ascorbate levels and increases protein nitration, a marker of oxidative stress [64]. These processes were accompanied by a decrease in the effective refractory period, changes consistent with atrial electrical remodeling [63]. Peroxynitrite and hydroxyl radicals, products of oxygen radical generation, have also been demonstrated in experimental models of AF. Experimental studies concluded that elevated levels of myeloperoxidase might favor the occurrence of AF in animals [64]. In experimental and clinical studies, a significant association between NADPH (nicotinamide adenine dinucleotide phosphate) oxidase-derived oxidative stress and AF was demonstrated [64].
Contrary to all these experimental data, in vivo studies have been difficult to perform because of the lack of reliable methods to quantify oxidative stress [65]. Oxidized glutathione and lipid peroxidation levels are elevated in coronary artery bypass surgery [65]. In patients after cardiac surgery, NADPH oxidase-derived ROS are overproduced in the right and left atria and are predictive of postoperative AF [63–65]. NADPH oxidase and dysfunctional nitric oxide synthase (NOS) contribute to superoxide production and may play an important role in atrial oxidative injury and electrophysiological remodeling in patients with AF [65].
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


