Fig. 26.1
With blue color all reports, with red color reports using animal models
26.2 What Has Been Done
The most frequently used animal models for studying cardiorenal syndrome are rodent models. The studies can be classified in
(a)
those focusing on cardiac alterations in models of chronic kidney disease usually caused by subtotal nephrectomy,
(b)
those focusing on renal alterations in models of heart failure usually caused by left anterior descending coronary artery ligation and
(c)
those (actually very few and largely inconclusive) focusing on combined cardiac and renal damage.
These studies and their cumulative results can be found tabulated in a recent excellent review by Bongarts et al. [5].
From these studies, we would like to review in detail two from the group of H. Krum, quite representative of the status of the field.
26.2.1 The Acute Heart Failure Model
In order to assess the role of myocardial infarction (MI) on renal anatomy and function (the type 1 cardiorenal syndrome according to the already presented classification), Lekawanvijit and colleagues [6] have used a rat model where the left anterior descending artery was ligated [7]; this method leads to an overall mortality rate of about 30 %. Animals (operated, sham-operated and controls) were sacrificed at several time intervals post-infarction, ranging from 1 to 16 weeks. Assessment of cardiac function demonstrated that animals with MI had a lower ejection fraction compared to the other groups; blood pressure was unchanged, except for the early (1 week) interval in the animals with MI. Systolic and diastolic dysfunction was documented for all time intervals in the infarct group.
Renal function was evaluated by measurement of Glomerular Filtration Rate (GFR) and albuminuria. GFR was decreased at early time points (1 and 4 weeks) but tended to reach normal levels in subsequent time intervals. Albumin in 24 h urine was normal till the 4-week interval and showed an increase at later time points, reaching statistical significance at 16 weeks. Half of the infarct animals exhibited focal tubulointerstitial scarring at 16 weeks; they all showed peritubular fibrosis in the cortex in a time-dependant fashion. The interstitial space of those animals also exhibited macrophage infiltration at the 1-week interval only; along the same lines, IL-6 mRNA was upregulated at the 1 week interval. The TGF-β mRNA, the protein itself and downstream effector phospho-SMAD-2 (a crucial for signaling transcription factor) were upregulated with different kinetics. Study of acute kidney injury biomarkers showed no change in NGAL (Neutrophil Gelatinase Associated Lipocalin) but a biphasic increase in KIM-1 (Kidney Injured Molecule 1) positive tubules.
The authors postulate that there may be a bi-phasic effect of myocardial infarction on renal structure and function: At an early phase, the decrease in ejection fraction and hypotension may lead to reduction in blood flow through the kidneys and reduction of GFR, resulting in hypoxic/ischemic injury. Following this phase and as a result of these early changes, profibrotic cytokines such as IL-6 and TGF-β are responsible for a gradual development of fibrosis in the renal parenchyma, leading to long-term anatomical changes and renal dysfunction. Contributors to this second phase may not be limited to the TGF-β pathways; although experimental evidence is lacking, it is possible that the rennin-angiotensin-aldosterone system and the endothelin system may be involved as well [8].
26.2.2 The Chronic Renal Failure Model
In order to evaluate the role of chronic kidney disease on cardiac function (the type 4 cardiorenal syndrome according to the proposed classification) Lekawanvijit and colleagues [9] have used the rat model of subtotal nephrectomy (STN). In this model, one kidney is removed and two-thirds of the blood supply of the other kidney are blocked. These interventions are followed by a substantial reduction of the functional renal mass, leading to fibrosis and chronic kidney disease. At different time intervals (4, 8 and 12 weeks), experimental and control animals were studied for the levels of the uremic toxin indoxyl sulfate, renal function, cardiac function, cardiac anatomy/fibrosis and for macromolecules with possible involvement in the alterations observed. Indoxyl Sulfate (Fig. 26.2) is a protein-bound, poorly dialyzable toxin; it is a derivative of tryptophan (tryptophan absorbed at the intestine is metabolized to indole and indole in the liver is metabolize to indoxyl sylfate). Indoxyl sulfate is normally excreted in the urine, but in CKD elevated serum levels are observed [10]. The reason for focusing on indoxyl sulfate was that previous findings in vitro have indicated that indoxyl sulfate exerts pro-inflammatory, pro-fibrotic and pro-hypertrophic effects on cardiac fibroblasts and cardiomyocytes [11].
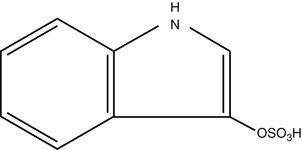
Fig. 26.2
Chemical formula of Indoxyl Sulfate
A consistent decrease in body weight, in the range of 10 % was observed in animals with STN. A significant increase in systolic blood pressure, in the range of 50 % was recorded at all time intervals. As expected, animals with STN had increased serum creatinine and urine total protein concentrations and reduced GFR. Also, regarding indoxyl sulfate, animals with STN had almost four times higher concentrations in serum and reduced concentrations in urine. Administration of an oral charcoal absorbent of indoxyl sulfate, kremezin (AST-120), had no effect on blood pressure or body weight, but could improve serum creatinine and urine total protein. The heart weight, the left ventricular weight and the lung weight, all adjusted to body weight, were increased in animals with STN; echocardiography established early diastolic dysfunction and cardiac hypertrophy. Cardiac tissue findings relate to inflammation, fibrosis and hypertrophy. The pro-inflammatory cytokines IL-6 and TNF-α were examined at the mRNA level and no difference was detected between sham-operated and STN animals. Cardiac fibrosis was studied both morphologically and biochemically. Extracellular matrix evaluation with Sirius Red staining demonstrated an extensive fibrosis, which was reduced 50 % in animals taking kremezin. Examination of genes and pathways related to fibrosis indicated that TGF-β was increased at the mRNA and the protein level in STN animals and signaling pathways involving NFκB, p38 and p44/42 MAKPs were activated; some of these phenomena were partly reversed by the use of kremezin. Regarding hypertrophy, it was observed that cardiomyocyte cross-sectional area was significantly increased in STN animals and pro-hypertrophic genes such as ANP, βMHC and αSMA were also upregulated; none of these phenomena was affected by administration of kremezin.
Based on these results it can be concluded that CKD-generated uremic toxin IS contributes to uremic cardiomyopathy by inducing cardiac fibrosis. Despite signaling defects in cardiomyocytes, detected with high levels of IS, the study did not clearly document a functional impairment in the heart. These findings, combined with previous studies using the IS absorbent kremezin, imply a beneficial effect of reducing this specific uremic toxin. However, the contributions of the rennin-angiotensin-aldosterone system should be also examined in this model, as acknowledged by the authors.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


