Cardiopulmonary Bypass and the Lung
David Kiamanesh
Alan Gaffney
Robert N. Sladen
INTRODUCTION
Cardiopulmonary bypass (CPB) consistently induces a wide range of acute lung injury, from mild to massive. Post-CPB pulmonary management is compounded by multiple factors that may compromise function, including atelectasis, pleural disruption, impaired lung compliance, capillary leak from an acute inflammatory response, and even fulminant acute respiratory distress syndrome (ARDS). To the patient who enters cardiac surgery with already limited pulmonary reserve, CPB represents a particular challenge. This chapter addresses the current state of knowledge of pulmonary function in the setting of CPB, and outlines therapeutic approaches to the management of the lungs during CPB.
CPB-INDUCED ATELECTASIS
Pathophysiology of Atelectasis
Anesthesia and Neuromuscular Blockade
Atelectasis is common after cardiac surgery and general anesthesia, and several mechanisms have been proposed to explain this phenomenon (1). After the induction of general anesthesia with neuromuscular blockade, mechanical ventilation initiates atelectasis within 5 minutes (2). The relaxed diaphragm is displaced cephalad by the abdominal contents. This increases pleural pressures and compresses adjacent lung tissue (3,4), and gas flow is preferentially distributed to the nondependent regions of the lung. This promotes ventilation-perfusion mismatch, hypoventilation, and progressive microatelectasis of the dependent lung zones. Without interruption by the intermittent “sighs” that occur in the awake patient, the monotonous mechanical ventilatory pattern during anesthesia allows the extension of atelectasis from alveoli to segments, lobules, and even lobes of the lung.
Resorption atelectasis is induced when capillary uptake of oxygen exceeds the rate of alveolar oxygen influx (5). Resorption may also occur in lung zones that have low ventilation/perfusion (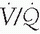 ) ratios; the uptake of oxygen increases significantly as the fraction of inspired oxygen (FIO2) increases (3).
) ratios; the uptake of oxygen increases significantly as the fraction of inspired oxygen (FIO2) increases (3).
) ratios; the uptake of oxygen increases significantly as the fraction of inspired oxygen (FIO2) increases (3).The proximate cause of pulmonary atelectasis during CPB is cessation of ventilation and lung collapse. There is some evidence that this may contribute to pulmonary dysfunction postoperatively (6,7). Resumption of ventilation prior to weaning from CPB is seldom fully successful in lung reexpansion. Following CPB, the patient is left with a variable degree of residual atelectasis, from microscopic to lobar in magnitude.
Surfactant Depletion
Pulmonary surfactant covers the alveolar surface and lowers surface tension to prevent alveolar and small-airway collapse. There is laboratory evidence that exposure to anesthesia impairs surfactant function and increases the permeability of the alveolar capillary barrier (8,9). Alterations in surfactant composition and function after CPB have been reported, but clinically significant changes in surfactant function have not been studied at length (10,11). Because of an extensive surfactant reserve and long turnover time, the role of surfactant depletion in the setting of CPB is unclear. Atelectasis itself promotes the production of proinflammatory cytokines, which decrease the synthesis of surfactant (12).
Anatomic Considerations
During CPB, the heart rests on the immobile left lower lobe. With blind bronchial suctioning, the catheter usually enters the more direct right main stem bronchus, resulting in preferential right bronchial drainage. Traumatic mucosal “pitting” induced by the suction catheter at the carina causes secretions to dam up and promotes airway collapse (13). When the pleural cavity is opened, blood and fluid can enter and compress the adjacent lung. In patients who undergo coronary revascularization, dissection of the left internal mammary artery mandates entry into the left pleural space. Together, these factors account for the 60% to 70% incidence of left lower lobe atelectasis after CPB (14).
The right lung can be compressed during cannulation of the inferior vena cava. The supine position is associated with a decrease in functional residual capacity (FRC) as abdominal contents push the diaphragm cephalad and promote small-airway closure. Significant atelectasis has been observed in the dorsal lung regions in patients after cardiac surgery (15).
High Inspired Oxygen Concentration
High inspired concentrations of oxygen (FIO2) during the perioperative period promote atelectasis (16,17,18). When the alveolar-arterial oxygen concentration gradient (AaDO2) increases, the capillaries rapidly absorb oxygen, resulting in alveolar atelectasis. The degree of atelectasis varies at different oxygen concentrations, and it is most notable when the FIO2 is >0.8 (19,20).
Low Tidal Volume Mechanical Ventilation
The use of low tidal volumes as a component of a protective lung strategy is now the standard of care in the setting of ARDS (21). Although this protects against alveolar stretch-induced injury (volutrauma), it also promotes atelectasis (atelectrauma). Pressure controlled ventilation (PCV) is a ventilator mode that limits peak airway pressures in order to decrease the risk of pulmonary barotrauma. However, if lung compliance is very poor, PCV may result in delivery of small tidal volumes that promote the development of atelectasis.
Comorbid Conditions
In heavy smokers with chronic bronchitis, the ciliated columnar epithelium undergoes metaplasia to mucin-producing goblet cells (22). Anterograde cilial clearance of mucus and debris is impaired, surfactant production is diminished, and small airways and alveoli tend to collapse.
Clinical Consequences of Atelectasis
It is common for the FRC to be decreased by as much as 20% for up to a week after general anesthesia. CPB-induced atelectasis causes more severe disruption of the broncho-alveolar tree and as a consequence FRC is decreased by as much as 40% to 50% (25). This causes a substantial impairment of lung compliance and promotes a pulmonary inflammatory response that contributes to postoperative pulmonary complications (26,27,28).
Impaired Oxygenation
The AaDo2 consistently increases after CPB (29,30,31,32,33). It increases to a maximum about 48 hours postoperatively, does not return to normal for at least 7 days, and is detectable for weeks after surgery (34). However, the increase in AaDO2 is similar between patients undergoing on-pump or off-pump coronary artery bypass grafting, suggesting that etiology is the surgical procedure itself, rather than CPB (35,36).
Intrapulmonary shunt (Qs/Qt) and veno-arterial admixture are consistently increased after CPB, but there is a poor correlation between increases in Qs/Qt and decreases in measured FRC (37,38). When airways collapse, Qs/Qt is worsened, secretion clearance is compromised, and the risk of pulmonary infection and pneumonia is increased. In animal models, induced alveolar collapse results in regional areas of hypoxemia that may reflexly increase pulmonary vascular resistance to the extent that right ventricular dysfunction develops (39).
Decreased Lung Compliance
The decline in lung compliance induced by atelectasis increases the work of breathing required for alveolar distention. A decrease in the expiratory reserve volume and FRC promotes further collapse because there is a lower volume remaining in the lung at the end of a normal tidal ventilation. As a consequence, the inspiratory and expiratory components of the breath duty cycle occur within a less efficient portion of the pressure-volume curve (40,41). If severe enough, this can lead to a substantial increase in energy consumption and acute ventilatory failure, with continued requirement for mechanical ventilation.
Acute Lung Injury
The repetitive opening of collapsing alveoli leads to surfactant inactivation and atelectrauma, and has been implicated in the development of ventilator-induced lung injury (VILI). When alveoli are stretched by excessively large tidal volumes, inflammatory mediators and neutrophils are activated, leading to increased alveolar capillary permeability and hyaline membrane formation. Atelectrauma may be prevented or attenuated by a protective lung strategy that limits tidal volumes to curtail inspiratory alveolar stretch and adds positive end-expiratory pressure (PEEP) to prevent expiratory alveolar collapse (21).
Prevention and Treatment of Perioperative Atelectasis
Strategies to prevent and treat atelectasis in patients undergoing CPB have focused on recruitment maneuvers and efforts to prevent de-recruitment of alveoli. The resultant modifications in mechanical ventilation may improve gas exchange, increase FRC, and prevent atelectasis-induced lung injury.
Continuous Positive Airway Pressure Ventilation
Static inflation of the lungs by continuous positive airway pressure (CPAP) during CPB has attracted considerable interest as a means of preventing CPB-induced atelectasis, with initial studies demonstrating benefit. One demonstrated that application of CPAP at 10 cmH2O during CPB improved postoperative arterial oxygen tension (PaO2), decreased AaDO2, and Qs/Qt for up to 4 hours after CPB and at the time of tracheal extubation (42), a response attenuated at lower levels of CPAP (43). It was suggested that the lung expansion induced by CPAP might preserve pulmonary mechanics by preservation of bronchial perfusion (44). Furthermore, it was considered that static inflation of the lungs with oxygen might prevent ultrastructural damage by sustaining cellular oxidative metabolism (45). In small animal studies, CPAP during CPB is associated with lower pulmonary lactate/pyruvate ratios and greater high-energy phosphate concentrations compared with
passive lung collapse (46). Several studies have demonstrated a decrease in inflammatory markers (47,48,49).
passive lung collapse (46). Several studies have demonstrated a decrease in inflammatory markers (47,48,49).
However, these findings have not been correlated with sustained postoperative benefit. A meta-analysis of eight studies published between 1990 and 2008 that included 460 patients reviewed the impact of CPAP, low-volume mechanical ventilation, and vital capacity maneuvers on arterial oxygenation, oxygenation index, AaDo2, and Qs/Qt (50). Although these maneuvers significantly improved these oxygenation parameters immediately after separation from CPB, the benefits were short-lived and had no apparent impact on clinical outcomes.
Recruitment Maneuvers
There is considerable evidence that sustained inspiratory recruitment maneuvers on CPB may inflate collapsed alveoli, and improve arterial oxygenation and lung compliance (3,6,51,52). However, peak inspiratory pressures of up to 40 cmH2O may be required. Although identical “Valsalva” maneuvers are routinely used after open-heart procedures to expel air from the left atrium and ventricle, there is a risk of disruption of the internal mammary graft by excessive distension of the left lung, or rupture of a bleb or weakened lung area. “Sighs” provided after separation from CPB can impede venous return and add to hemodynamic instability.
The “Open Lung” Concept
The recruitment maneuver has been extrapolated to the socalled open-lung concept, which requires an initial period of PCV with high airway pressures (40 cmH2O) with inverted inspiratory:expiratory (I:E) ratio from 1:2 to 1:1. This is followed by a protective lung strategy with low tidal volumes and PEEP to minimize atelectrauma. Application of the open-lung concept to cardiac surgery has minimized post-CPB atelectasis, improved FRC, and decreased the incidence of postoperative hypoxemia (53). Moreover, when the open-lung concept is commenced immediately after anesthetic induction and tracheal intubation, the inflammatory response to CPB is attenuated, with decreased levels of the proinflammatory interleukins IL-8 and IL-10 (54).
CPB AND PULMONARY MECHANICS
There is considerable evidence that CPB has a deleterious effect on pulmonary mechanics (31,55,56,57,58,59), but it may be difficult to distinguish these from those induced by thoracotomy, pleural resection, and pleural effusions. Decreases in vital capacity, FRC, lung compliance, and pulmonary function have been shown to persist for more than 3 months after surgery (60,61,62,63), although in several studies the alterations have been considered to have a negligible clinical impact (64,65,66).
To resolve the question of the role of CPB versus cardiac surgery itself on pulmonary mechanics, trials have compared on-pump and off-pump coronary artery bypass surgery. In one prospective randomized trial of 197 cardiac surgery patients, OPCAB surgery was associated with improved oxygenation and decreased time to tracheal extubation. However, there were no differences in chest radiographs, spirometry, and incidence of pneumonia, pleural effusion, pulmonary edema, or mortality between groups (67). Although these beneficial effects have been confirmed by some (36,68), others have found no difference in pulmonary mechanics or oxygenation (69,70). In fact, a smaller randomized study of 35 patients paradoxically found a worse pulmonary shunt in patients after OPCAB surgery despite a partial attenuation of the inflammatory response (71). Benefits have been observed by the avoidance of CPB in patients with chronic obstructive pulmonary disease, in whom OPCAB or minimally invasive approaches are associated with decreased atelectasis, duration of tracheal intubation, and intensive care unit length of stay (72).
Airway resistance is consistently increased by CPB, and is more pronounced than in patients undergoing OPCAB surgery (73). There have been a number of reports of fulminant bronchospasm during CPB (74,75,76,77). It is most likely related to contact activation with an idiosyncratic excess activation of human C’5a anaphylatoxin, although other causes of such bronchospasm, for example acute non-cardiogenic pulmonary edema (“cardiac asthma”), must not be overlooked. (78).
Operative technique and graft selection can also alter pulmonary mechanics. Dissection of the internal mammary artery with violation of the anterior chest wall and pleural cavity decreases FRC and forced expiratory volume in 1 second (FEV1) to a greater extent than when saphenous vein grafts are used alone (79,80). Lung retraction and pleurotomy, rather than CPB itself, are major architects of disturbed lung mechanics during cardiac surgery (31,55).
CPB-INDUCED ACUTE LUNG INJURY
Pathophysiology of Acute Lung Injury
Acute lung injury with fulminant noncardiogenic pulmonary edema is a rare but life-threatening complication of CPB (81,82,83,84,85). After the advent of CPB in the 1950s, a substantial proportion of deaths following cardiac surgery occurred consequent to a syndrome of acute respiratory failure referred to as “pump lung.” Its morphology is identical to that of ARDS associated with other inflammatory syndromes such as sepsis (86). There is diffuse lung injury and swelling of endothelial cells and pneumocytes, with an uneven distribution of surfactant (87). Broncho-alveolar lavage (BAL) after CPB demonstrates an increase in IL-8 levels and neutrophil percentage (88).
Noncardiogenic pulmonary edema results from increased permeability of the alveolar capillary membrane, creating a capillary leak syndrome with exudation of water and protein into the alveolar space. It is distinguished from cardiogenic pulmonary edema by the finding of normal or low left atrial or pulmonary artery wedge pressures, and a high protein concentration in the edema fluid (albumin concentration ≥90%
of serum albumin). In its most dramatic form, it is heralded by a massive outpouring of frothy, proteinaceous fluid from the tracheal tube. Fluid loss may culminate in intravascular volume depletion and hypovolemic shock, with death resulting from low cardiac output syndrome rather than hypoxemia (83). Thus, even in patients with diffuse pulmonary edema and severe hypoxemia, aggressive restoration of intravascular volume is mandatory, guided by close monitoring of left ventricular filling pressure.
of serum albumin). In its most dramatic form, it is heralded by a massive outpouring of frothy, proteinaceous fluid from the tracheal tube. Fluid loss may culminate in intravascular volume depletion and hypovolemic shock, with death resulting from low cardiac output syndrome rather than hypoxemia (83). Thus, even in patients with diffuse pulmonary edema and severe hypoxemia, aggressive restoration of intravascular volume is mandatory, guided by close monitoring of left ventricular filling pressure.
The Role of CPB in Acute Lung Injury
It was initially assumed that acute lung injury during CPB is triggered by embolism of particulate debris into the pulmonary microcirculation. This in turn evokes an inflammatory response mediated by complement activation, neutrophil migration, cytokine production, and arachidonic acid metabolites. However, it soon became apparent that “pump lung” occurs even when all the perfusate is filtered, suggesting a role for etiologies in addition to or instead of microembolism. For example, the incidence of postoperative respiratory complications is not significantly different in OPCAB surgery (70,89). The lung inflammatory response may be as much an epiphenomenon of surgery on the heart, and its incidence may be related to operator technique. Other factors also contribute. For example, high FIO2 in the early postoperative period increases cytokine levels and augments the proinflammatory response of alveolar macrophages (90).
Contact Activation and the Inflammatory Cascade
Complement activation and the potential for pulmonary dysfunction appear to be a consistent response to contact activation of blood in extracorporeal circuits, such as leukophoresis and hemodialysis (91,92). Contact activation has not been demonstrated in OPCAB surgery (93).
Contact activation of the blood by the extracorporeal circuit triggers a series of amplification cascades mediated by proteolytic enzymes, particularly serine proteases. It is initiated by the activation of the circulating procoagulant, factor XII (Hageman factor), which triggers the intrinsic pathway of coagulation, fibrinolysis, and complement (C’3a and C’5a) activation via the classical and alternate pathways. C’5a has spasmogenic and leukocyte-activating properties (78). When C’3a, the central component of the system, is cleaved, the fragments that result are called anaphylatoxins. These induce smooth muscle contraction and function as a major chemotactic factor for neutrophils. They trigger the release of arachidonic acid metabolites such as leukotriene B4, which increases vascular permeability and provokes mast cell degranulation, and induce the formation of neutrophil adhesion molecules. Higher expression of adhesion molecules after CPB is seen in patients with diabetes and may increase the risk of postoperative complications (94).
The incidence and severity of postoperative organ dysfunction correlates with plasma complement concentrations, duration of CPB, and the degree of elevation of C’3a levels (95). Complement-exposed neutrophils become “sticky”, adhere to surfaces, aggregate, and may block the microcirculation by margination and leukoembolization. The neutrophils degranulate and release highly reactive oxygen free radicals and proteolytic enzymes, which damage endothelial cells. The net effect is a dramatic increase in vascular permeability and capillary leak syndrome.
Neutrophil Activation
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


