Cardiomyopathy
“If it takes more than 10 minutes of discussion to decide if a patient needs an operation, it is best not to operate”
There are three distinct cardiomyopathies: Hypertrophic, dilated or congestive, and restrictive. Recently, a fourth type, “ventricular noncompaction” has been described.
Hypertrophic Cardiomyopathy
Hypertrophic cardiomyopathy (HC) is a condition in which myocardial thickness is increased. In general, the septal thickness is more marked than that of the posterior wall (see Fig. 13-1). There may or may not be left ventricular (and in some cases, right ventricular) outflow tract obstruction. If there is left ventricular outflow tract obstruction, the condition is termed hypertrophic obstructive cardiomyopathy (HOCM). If there is no left ventricular outflow tract obstruction, it is termed hypertrophic nonobstructive cardiomyopathy. When there is left ventricular outflow tract obstruction usually there is systolic anterior motion of the mitral valve apparent on echocardiography. Mitral insufficiency frequently can be associated with HC. HC can be associated with chest pain, shortness of breath, exercise intolerance, syncope, and sudden death. It is the most common identifiable nontraumatic cause of death on the athletic field.
Etiology and Genetics
Left ventricular septal and posterior wall hypertrophy can result from numerous causes. However the terms obstructive and nonobstructive HC usually imply an autosomal dominant condition that results from mutations in one of the following genes that encode proteins of the cardiac sarcomere: β-Myosin heavy chain, cardiac troponin T, α-tropomyosin, and myosin-binding protein C genes, as well as in two genes encoding the myosin light chains. As more causative genes are discovered, the taxonomy for what might now be considered the “wastebasket” term “HC” will change completely. This will be necessary because knowledge of the specific gene defect will allow better definition of the natural history of the disease.
Therefore, when faced with a patient who is known (usually because of an echocardiogram) to have abnormally thick left ventricular walls one must ascertain the cause of
the ventricular hypertrophy. This is particularly important for infants and young children because some believe that in these age-groups one is more likely to find underlying causes for the hypertrophy than in older age-groups.
the ventricular hypertrophy. This is particularly important for infants and young children because some believe that in these age-groups one is more likely to find underlying causes for the hypertrophy than in older age-groups.
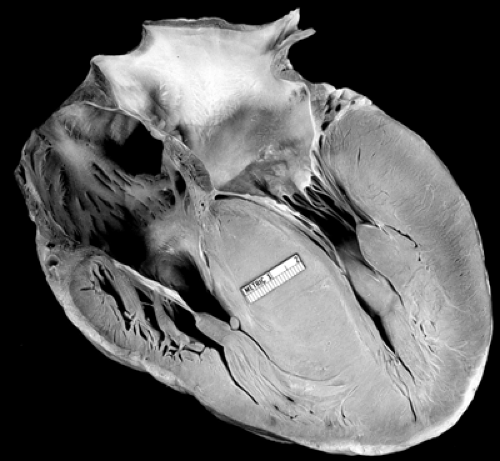 Figure 13.1 • Pathologic specimen of hypertrophic cardiomyopathy. Note the markedly thickened septum and left ventricular posterior wall. |
In the newborn period, it is well-recognized that infants born to mothers with diabetes can have a transient form of HC. Other conditions that can be associated with increased left ventricular wall thickness are glycogen storage diseases, mitochondrial disorders, disorders of oxidative metabolism, carnitine deficiency, β-oxidation defects, Noonan syndrome, Costello syndrome, Friedreich’s ataxia, nonspecific causes of cardiac inflammation, and steroid administration among others. Once all of these causes have been eliminated, one is left with a diagnosis of HC presumably resulting from sarcomere mutations, which, with the current state of knowledge, are lumped together for purposes of ascertaining the natural history and the results of treatment.
Once a diagnosis of a presumed sarcomeric form of HC is made, first-degree relatives of the index cases should undergo echocardiograms to screen for familial cases. For first-degree relatives with normal echocardiograms, the interval between subsequent screenings is unclear. Some investigators have suggested yearly screening for children and adolescents on the assumption that HC is more likely to become apparent during the years of rapid growth. Other investigators think that screening echocardiograms every 2 to 5 years is reasonable. Gene testing to identify affected family members is becoming available. Initially, one needs to identify the gene mutation in the patient and then screen the family members for that gene.
History
Patients with HC usually come to medical attention in one or more of the following ways: Detection of a murmur, an abnormal electrocardiogram, from family screening or a positive family history, from evaluation of syncope, chest pain, palpitations, or out of hospital cardiac arrest.
Physical Examination
The physical findings can be variable. For patients with nonobstructive forms of HC, there is no characteristic murmur. There may or may not be a prominent apical impulse, and an S4.
In some patients, obstruction can be unmasked and a murmur will appear with maneuvers to lower systemic vascular resistance such as inhalation of amyl nitrate or assuming a standing position.
In some patients, obstruction can be unmasked and a murmur will appear with maneuvers to lower systemic vascular resistance such as inhalation of amyl nitrate or assuming a standing position.
Patients with obstructive HC will have systolic ejection murmur. Unfortunately, in subtle cases one can mistake this murmur for an innocent flow murmur. There may be a bifid pulse.
Electrocardiogram
The classic electrocardiogram shows evidence of left and frequently biventricular hypertrophy. For patients with HC and restrictive left ventricular filling, there can be evidence of atrial enlargement.
Chest x-ray
The chest x-ray is not diagnostic.
Echocardiography
The echocardiogram is most important in making the diagnosis of HC. Classically, there will be asymmetric septal hypertrophy and systolic anterior motion of the mitral valve. Echocardiography and Doppler sonography will allow assessment of the absence, presence, and degree of left and right ventricular outflow tract obstruction. Echocardiography allows quantification of the thickness of the ventricular walls.
Natural History and Treatment
Although HC can be associated with symptoms of fatigue, shortness of breath, chest pain, and palpitations, the most important concern is the increased risk of sudden death. The exact magnitude of the risk of sudden death is still being clarified. In the past, it was thought that the risk of sudden death was 3% per year. However, more recent population-based studies indicate that the risk is closer to 1% per year. Several years ago, it was thought that one could predict patients who were at high risk of death by the specific gene defect causing the HC. Unfortunately, this has not proved to be correct. However, as more genes are identified and larger cohorts of patients with specific gene mutations are identified, this may yet be the case.
The ability to identify patients at high risk for sudden death is still crude but the following features may be associated with higher risk for sudden death: Diagnosis in childhood, septal thickness exceeding 30 mm, nonsustained ventricular tachycardia, failure of normal increase of systolic blood pressure during exercise, family history of premature death associated with HC, significant left ventricular outflow tract obstruction, and prior cardiac arrest.
The current paradigm for treating HC is to identify those patients at high risk for sudden death and implant an automatic internal cardiac defibrillator (AICD). There is reasonably good data that this strategy prolongs life. However, it is not clear which patients are at high enough risk to have this rather expensive treatment.
For symptomatic patients who are not at high risk for sudden death and do not qualify for implantation of an AICD, treatment with a β-blocker or amiodarone can be considered.
For patients with significant left ventricular outflow tract obstruction, surgical myotomy myectomy, or alcohol ablation can be done. There is increasing evidence that elimination of the obstruction prolongs life and relieves symptoms.
Patients with HC should be restricted from competitive athletics.
Dilated or Congestive Cardiomyopathy
Dilated or congestive cardiomyopathy is a descriptive diagnosis implying that the left ventricle is dilated with reduced systolic function (see Fig. 13-2). Normal left ventricular ejection fraction is 50% to 60%. Anything <50% is abnormal and could indicate the presence of dilated cardiomyopathy.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


