, Domenico Corrado2 and Cristina Basso1
(1)
Cardiovascular Pathology Department of Cardiac, Thoracic and Vascular Sciences, University of Padua, Padova, Italy
(2)
Cardiology Department of Cardiac, Thoracic and Vascular Sciences, University of Padua, Padova, Italy
An abrupt electrical turmoil precipitating ventricular fibrillation and cardiac arrest is a frequent complication of cardiomyopathies, whether overt or concealed, because they have an arrhythmogenic myocardial substrate.
When dealing with cardiomyopathies at risk of SCD, according to the World Health Organization, American Heart Association, and European Society of Cardiology definition of cardiomyopathies [1–3], we refer mostly to hypertrophic (HCM) and arrhythmogenic (AC) cardiomyopathies, which are often compatible with normal mechanical function, which is essential for sport activity, but are quite vulnerable in terms of electrical stability. Dilated and restrictive cardiomyopathies unlikely cope with effort performance, because of dyspnea and fatigue, and are an exceptional cause of unexpected SCD in the young and athletes.
4.1 Hypertrophic Cardiomyopathy
It is a major cause of SCD during effort and thus the leading cause of cardiac arrest in athletes in countries such as the United States, where ECG is not employed at the pre-participation screening [4–6]. It is a genetically determined disease, with autosomal dominant pattern of inheritance, mostly due to mutations of genes encoding sarcomeric proteins, and characterized by asymmetrical hypertrophy of the left ventricle, usually septal or apical [7, 8] (Figs. 4.1 and 4.2). Concentric, symmetrical hypertrophy may be also observed.
In case of asymmetrical septal hypertrophy, a fibrous endocardial plaque is usually present in the subaortic position, as a consequence of the friction of the anterior mitral leaflet with the endocardium of the ventricular septum, due to systolic anterior motion of the leaflet, which itself appears thickened (Figs. 4.2 and 4.3).
The histology discloses myocardial disarray, with variously oriented cardiomyocytes crossing each other (either single cell or fascicular), cardiomyocyte hypertrophy with bizarre nuclei, interstitial fibrosis, and dysplastic and frequently obstructed, intramural small arteries [9–16] (Fig. 4.4).
There are several structural substrates which explain why HCM is highly arrhythmogenic and at risk of life-threatening ventricular arrhythmias and SCD:
1.
Hypertrophy, which may be so huge as the heart can weigh up to 1000 g. Wall thickness greater than 30 mm is considered one of the major risk factors of SCD.
2.
3.
Fibrous scars, ischemic in origin, mostly due to impaired coronary flow reserve because of cardiac hypertrophy and myocardial restrictivity, with compression of small intramural coronary arteries during the diastole [15] (Figs. 4.5, 4.6, and 4.7); organic small vessel obstructive disease may also contribute (Fig. 4.4).
4.
Observation of scars within the hypertrophy is almost regular at autopsy of cases with HCM and SCD, even when the wall thickness and the heart weight are not particularly increased [16]. In support of the ischemic etiology of replacement-type fibrosis, all the stages of ischemic myocardial injury are seen in the hearts of people dying suddenly with HCM, including interstitial edema, myocyte coagulative necrosis, neutrophilic infiltrates, and myocytolysis, eventually leading to scarring [15] (Fig. 4.10). By the way, the progressive scarring accounts for the evolution of HCM into a dilated, end-stage form [17].
Disarray and replacement-type fibrosis appear to be a malignant arrhythmic combination. Replacement-type fibrosis is easily identified in vivo by contrast-enhanced cardiac magnetic resonance imaging [18].
A so-called idiopathic left ventricular hypertrophy has been reported in young people and athletes who died suddenly [4–6]. The degree of hypertrophy is beyond that seen in the cardiac hypertrophy of the trained athlete, with left ventricular thickness exceeding 15 or 16 mm. It is a diagnosis by exclusion, after ruling out other conditions that would predispose to left ventricular hypertrophy (e.g., aortic valve stenosis, isthmic aortic coarctation, systemic hypertension). Unlike HCM, idiopathic left ventricular hypertrophy is concentric and symmetric and there is no evidence of subaortic plaque and mitral valve disease, myocardial disarray, and familial transmission. Whether this condition should be considered a distinct entity at risk of SCD is still a matter of debate, and differential diagnosis with “athlete heart” remains a challenge not only in the clinical setting but also at postmortem [19–21].
4.2 Arrhythmogenic Cardiomyopathy
It is the leading nonischemic cause of SCD in the young and in the athletes in Italy [22, 23]. In contrast with HCM, which is a sarcomeric disease mostly affecting the left ventricle, AC is a desmosomal disease and has been viewed as a disease of the right ventricle [22–30]. The fibrofatty replacement starts from the subepicardium and deepens as a wave-front phenomenon, reaching the subendocardium, to become transmural [22, 31, 32]. In SCD series, both segmental (Figs. 4.11 and 4.12) and diffuse (Figs. 4.13 and 4.14) forms of AC are described and even cases with early stages of myocardial injury (Fig. 4.15), preceding the mature fibrofatty tissue [22, 24, 31–35]. Biventricular or even isolated or dominant left ventricular forms have also been reported [31, 32, 35–37] (Figs. 4.16 and 4.17). Left ventricular free wall involvement has been observed in up to 70 % of autopsy reports [31, 32], whereas the ventricular septum only in 20 % [31]. In the left ventricle, the subepicardium of the posterolateral wall is typically affected. When the biventricular involvement is diffuse, AC may mimic dilated cardiomyopathy with congestive heart failure, so severe as to require cardiac transplantation. Cases with predominant fatty tissue replacement with preserved wall thickness or even pseudo-hypertrophy do also exist (Fig. 4.18). However, isolated fatty tissue at postmortem cannot be regarded as a pathognomonic feature of the disease [19, 38], in order to avoid a misdiagnosis of AC at autopsy, with forensic and preventive implications, since the postmortem diagnosis must guide cascade investigation of family members [39].
Endomyocardial biopsy is frequently employed to detect fibrofatty replacement in vivo and an amount of residual myocardium less than 60 % is considered diagnostic [40]. Clinical diagnosis is challenging and there is no single gold standard [41]. Typically, the ECG discloses depolarization abnormalities, QRS widening, epsilon waves, and repolarization changes with inverted T waves on precordial leads. In the classical variant, ventricular arrhythmias present a left bundle branch block morphology indicating the right ventricular origin. Low voltage areas by electro-anatomic mapping and late enhancement by contrast cardiac magnetic resonance are now very helpful clinical imaging surrogates of myocardial atrophy [42, 43].
The pathobiological phenomenon consists of a transmural fibrofatty replacement of the ventricular free wall, as a repair consequence of a progressive cardiomyocyte death, either by necrosis or apoptosis [44, 45] (Fig. 4.19). In advanced forms, the myocardium may almost disappear and the free wall looks parchment like. This process accounts for the dilatation of the ventricular cavity and the development of wall aneurysms. In contrast with HCM, where the cardiomyocyte death is mostly ischemic in origin because of impairment of microcirculation due to hypertrophy and wall stiffness constricting the small arteries, in AC, cell death with myocardial loss is genetically determined, due to cell junction vulnerability [44]. AC is indeed a genetic disorder of desmosomes [24–30, 46, 47]. Mutations of genes encoding desmosomal proteins account for a vulnerable intercalated disk, which appears disrupted at electron microscopy [48, 49]. Stretch, due to increased ventricular preload during effort, facilitates both cell death and onset of arrhythmias. Although AC is genetically determined, there is an age and gender related penetrance of the phenotype and SCD typically occurs during adolescence or early adulthood, even as first manifestation of the disease [24–26, 50, 51].
The following substrates of the disease should be considered arrhythmogenic [51]:
1.
Fibrofatty replacement of the myocardium, which explains the QRS widening and postexcitation epsilon wave as well as inverted T waves in precordial leads at the ECG. Like in ischemic scars, fibrofatty replacement hinders and delays the intraventricular electrical impulse transmission, thus facilitating reentry phenomena.
2.
Development of aneurysms in the right ventricular free wall, which further favors reentry mechanisms for onset of ventricular arrhythmias of left bundle branch block morphology.
3.
Dilatation of the right ventricular cavity, especially with chamber overload during effort, with further slowdown of the intraventricular conduction.
4.
Inflammatory noninfectious infiltrates (“myocarditis”), most probably as a reaction to spontaneous cardiomyocyte death (Fig. 4.19). They have been reported at histology in up to 75 % of autopsy cases [31, 44, 52]. Bursts of cardiomyocyte necrosis with reactive inflammation may trigger life-threatening arrhythmias. This is most probably the explanation why they are so frequently observed at postmortem in subjects who had died suddenly.
It is still a matter of debate whether electrical instability at risk of SCD might occur also in the pre-phenotypic stage of the disease, before structural abnormalities of the myocardium, due to a cross-talk between mechanical junction and ion channels. Experimental studies demonstrated that intercellular space widening at the level of the intercalated disk and a concomitant reduction in action potential upstroke velocity, as a consequence of lower sodium current density, lead to slowed conduction and increased arrhythmia susceptibility at disease stages preceding the onset of necrosis and replacement fibrosis [49]. However, no single SCD case has been reported so far in the pre-phenotypic stage of AC.
4.3 Image Gallery
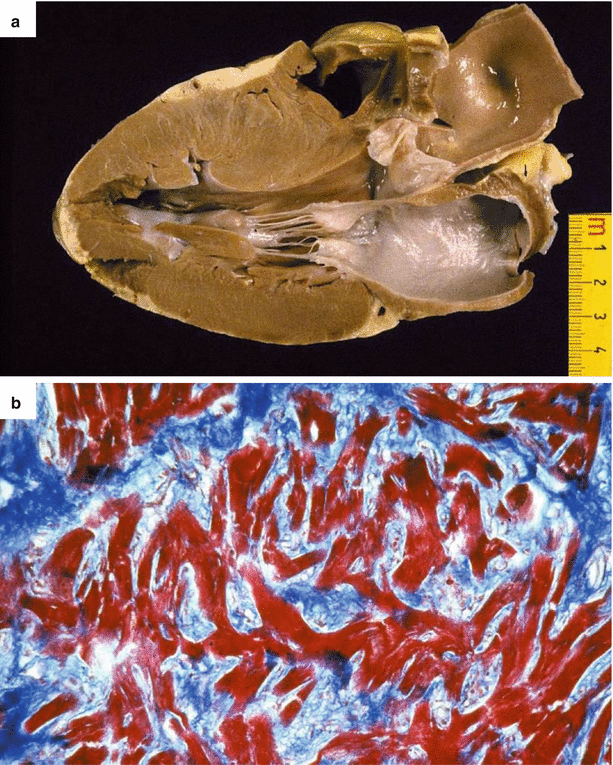
Fig. 4.1
Arrhythmic sudden cardiac death due to hypertrophic cardiomyopathy in a 37-year-old man during exercise. (a) Parasternal long-axis section of the heart shows asymmetric septal hypertrophy as compared to the posterior left ventricular wall. Note the absence of endocardial fibrous plaque. (b) At histology, extensive myocardial disarray and interstitial fibrosis (Heidenhain trichrome)
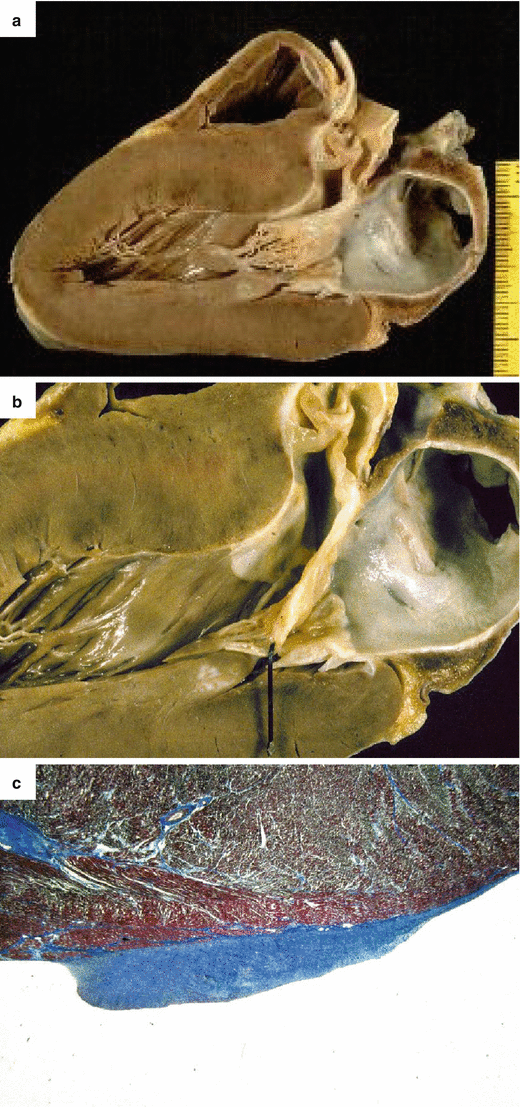
Fig. 4.2
Arrhythmic sudden cardiac death on effort due to hypertrophic cardiomyopathy in a 17-year-old athlete. (a) Parasternal long-axis section of the heart shows asymmetric septal hypertrophy with endocardial fibrous plaque. (b) Close-up of a: note the whitish endocardial plaque and the anterior mitral valve leaflet thickening. (c) Histology of the friction lesion showing endocardial fibrous thickening (Heidenhain trichrome)
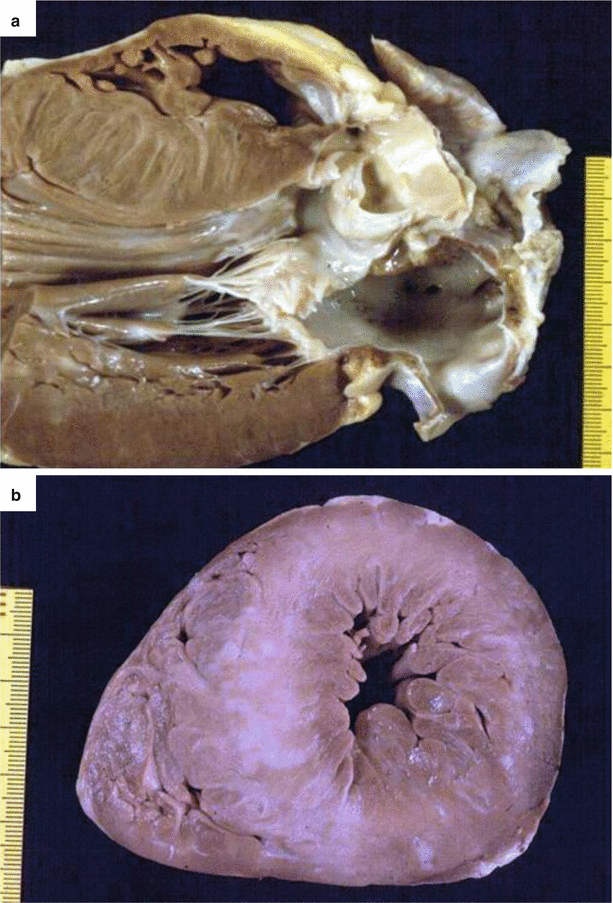
Fig. 4.3
Arrhythmic sudden cardiac death at rest due to hypertrophic cardiomyopathy in a 23-year-old man, with previous syncopal episodes and an in vivo diagnosis. (a) Parasternal long-axis section of the heart shows asymmetric septal hypertrophy (septal thickness about 30 mm) with endocardial fibrous plaque. (b) Short-axis view of the heart, showing massive left ventricular hypertrophy, with multiple whitish septal scars
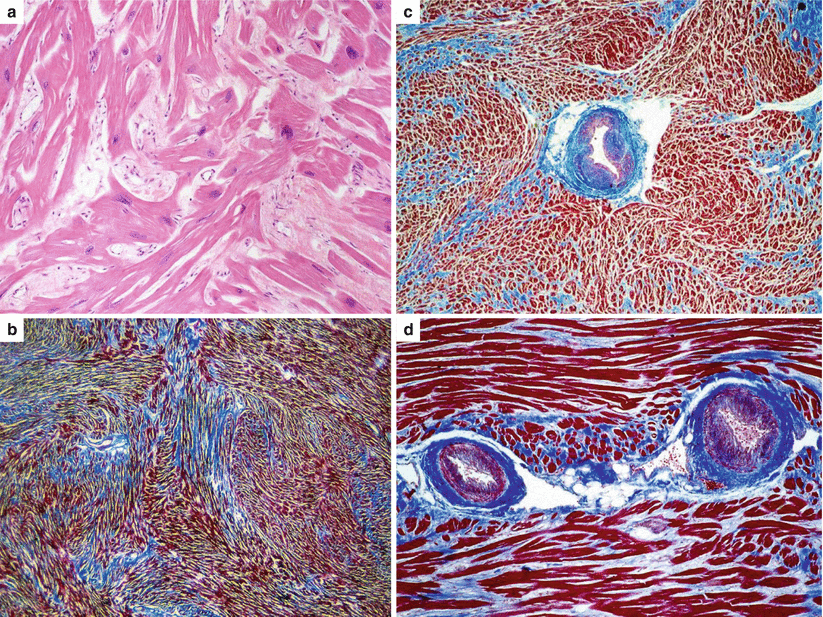
Fig. 4.4
Histological features of hypertrophic cardiomyopathy in sudden cardiac death victims. (a) Cardiac muscle cell disorganization (disarray), (hematoxylin–eosin). (b) Widespread fascicular myocardial disarray and interstitial fibrosis (Heidenhain trichrome). (c, d) Intramural small vessel disease (Heidenhain trichrome)
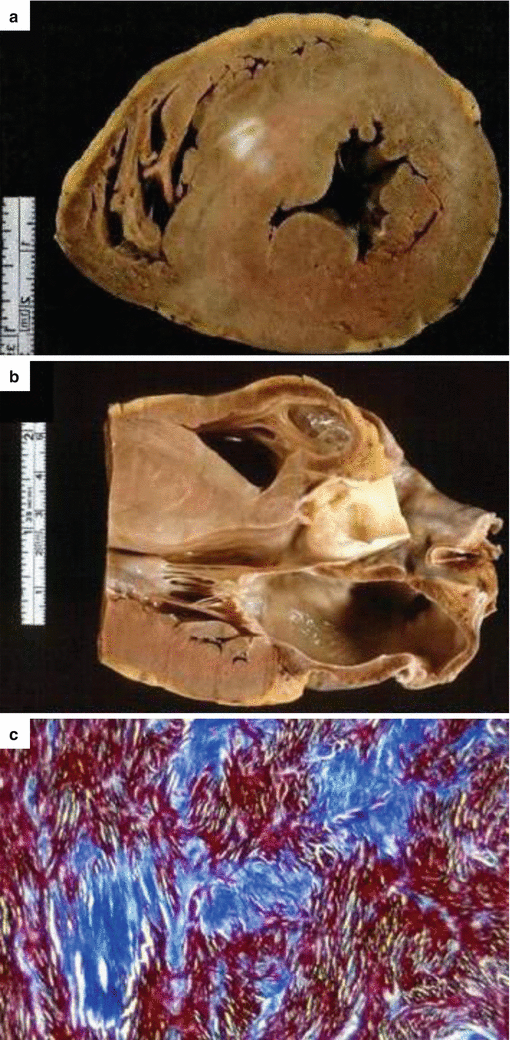
Fig. 4.5
Arrhythmic sudden cardiac death on effort due to hypertrophic cardiomyopathy in an 18-year-old soldier. (a) Cross section of the heart showing asymmetric septal hypertrophy and myocardial scars, (b) Parasternal long-axis section of the same. (c) Histology shows myocardial disarray, with replacement-type fibrosis (Heidenhain trichrome)
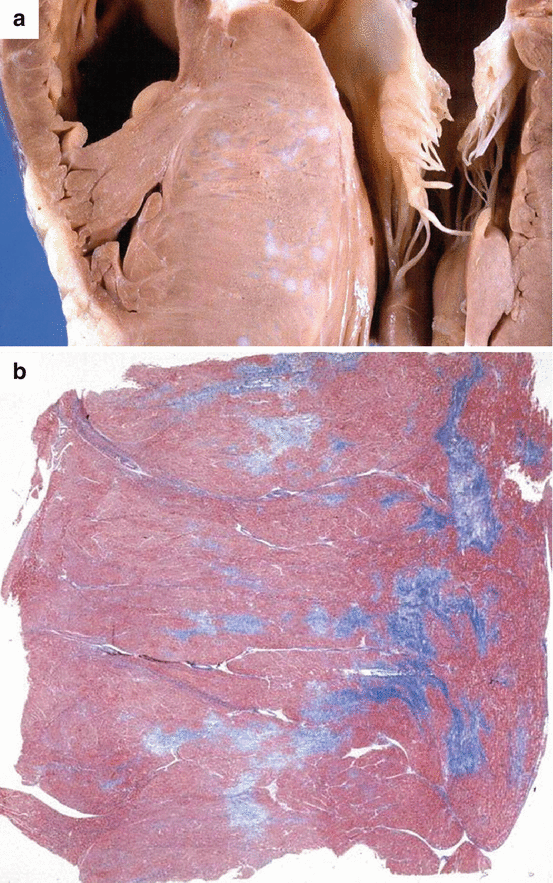
Fig. 4.6
Arrhythmic sudden cardiac death at rest due to hypertrophic cardiomyopathy in a 31-year-old man. (a) Parasternal longitudinal section of the heart showing asymmetric septal hypertrophy and myocardial scars. (b) Corresponding panoramic histological section showing multiple arcipelago-like areas of replacement-type fibrosis (Heidenhain trichrome)
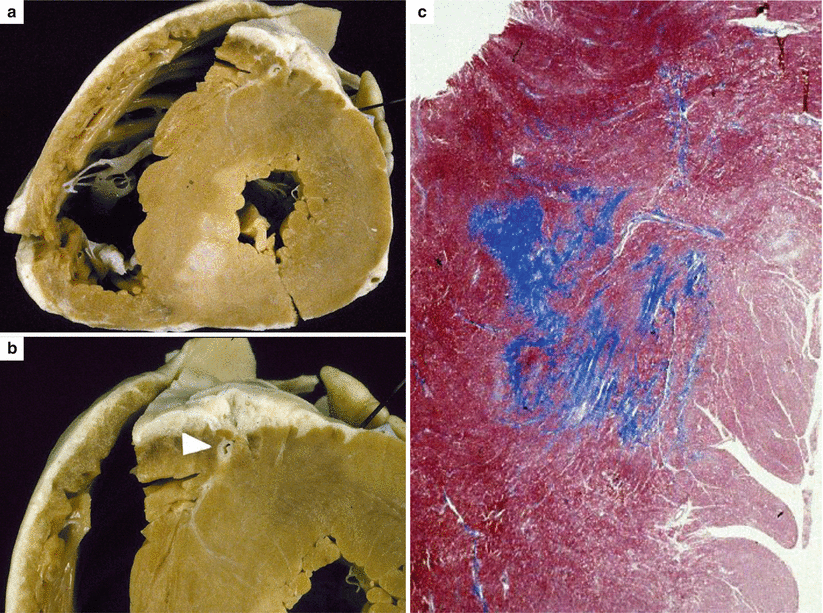
Fig. 4.7
Arrhythmic sudden cardiac death on effort due to hypertrophic cardiomyopathy in 15-year-old, previously asymptomatic boy, with a family history of the disease (father). (a) Short-axis view of the heart, showing massive concentric left ventricular hypertrophy and a large white scar in the posteroseptal area. (b) Intramyocardial course of the left anterior descending coronary artery (arrowhead). (c) Panoramic histological section of the septal area showing replacement-type fibrosis (Heidenhain trichrome)
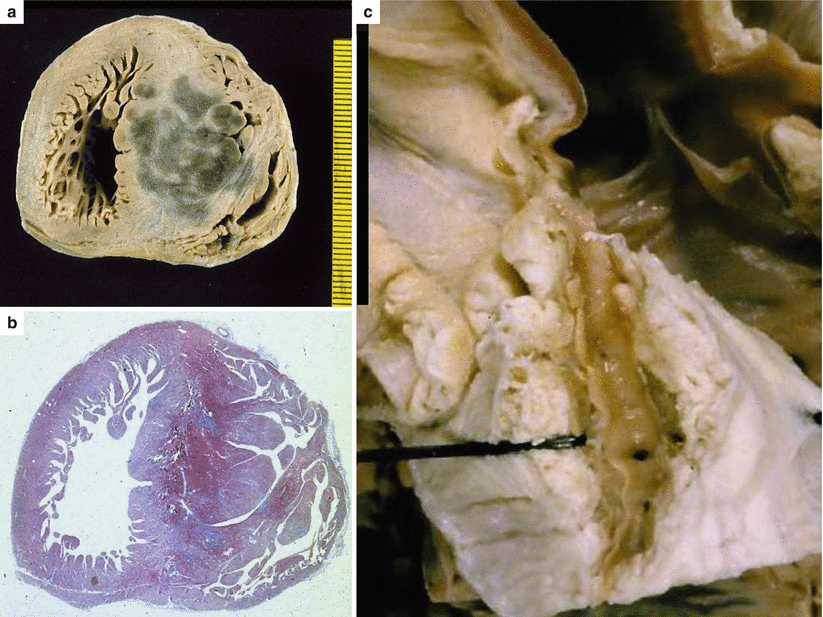
Fig. 4.8
Arrhythmic sudden cardiac death at school, due to hypertrophic cardiomyopathy in a 8-year-old asymptomatic boy. (a) Postmortem investigation reveales cardiomegaly with marked asymmetric septal hypertrophy (25-mm septal thickness vs. 7-mm thickness of the left ventricular free wall) and a hemorrhagic acute septal myocardial infarction. (b) Histological assessment revealed coagulation necrosis with interstitial hemorrhage as well as areas of replacement-type fibrosis (Heidenhain trichrome). (c) An intramural course of the left anterior descending coronary artery (2-mm-deep and 20-mm-long), corresponding to the region of the first and second septal branches, is visible
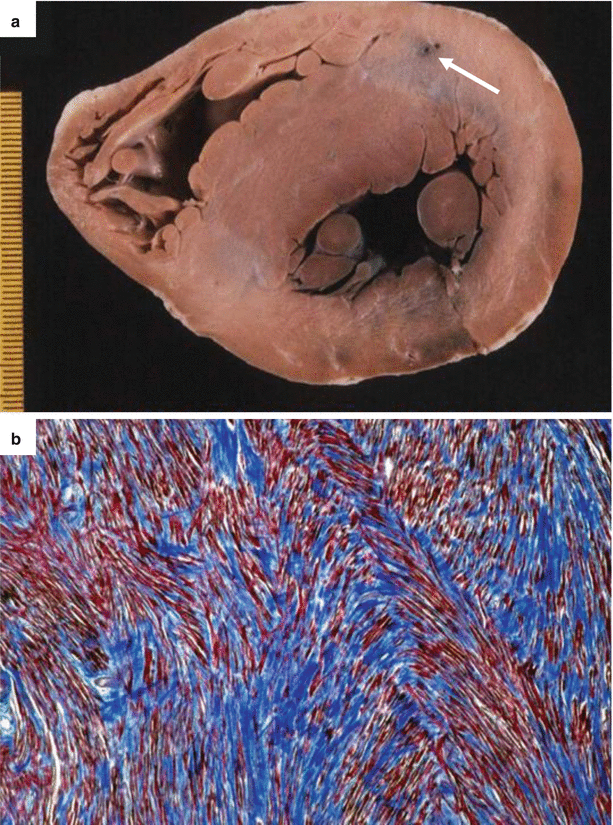
Fig. 4.9
Arrhythmic sudden cardiac death due to hypertrophic cardiomyopathy in a 29-year-old woman during jogging. (a) Transverse section of the heart specimen: note the absence of grossly visible fibrous scars and the deep intramural course of the left anterior descending coronary artery (arrow, depth 8 mm). (b) Marked myocyte disarray with tiny interstitial fibrosis (Heidenhain trichrome)
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


