Component
Constituents
Myofilaments
Actin, tropomyosin, troponin-C/I/T
Myosin
Hemisarcomere
Titin, nebulin,
MyBPc3
Z disc
α-actinin,
Tcap (telethonin), CRSP3 (MLP)
M line, H zone
Myomesin
Each thick filament in striated myocytes is constituted by two myosin heavy chains (MyHC) and four myosin light chains, more precisely, two pairs of essential (eMyLC or MyLC1) and regulatory (rMyLC or MyLC2) myosin light chains connected to the two heads of a given myosin filament (Vol. 5, Chap. 5. Cardiomyocytes).
The myosin heavy chain contains the actin- and ATP-binding sites. It thus represents the molecular nanomotor of muscle contraction. In the human heart, two MYH genes (MYH6 [αMyHC] and MYH7 [βMyHC]) are expressed. α-myosin heavy chain has a higher ATPase activity and shortening velocity than β-myosin heavy chain. The nanomotor is regulated by two myosin light chain (MyLC) isoforms. The MyLC1 subtype acts as a myosin heavy chain–actin tether. The MyLC2 isoform slows the rate of tension development of myosin [322]. The inhibition conferred by MyLC2 is relieved upon MyLC2 phosphorylation by myosin light chain kinase MLCK2. Expression of the atrial MyLC1 isoform in the hypertrophied human ventricle increases cross-bridge cycling and contractility [322].
In smooth myocytes and nonmuscle cells, phosphorylation of regulatory myosin light chains by MLCK switches on the actin-activated myosin ATPase, hence triggering contraction. In striated myocytes (i.e., skeletal and cardiac myocytes), the actin–myosin interaction is mainly regulated by the troponin–tropomyosin complex. Electrochemical stimulation increases the level of Ca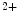 bound to the troponin–tropomyosin complex. Regulatory myosin light chains modulate the rate and magnitude of contractility, as MyLC2 phosphorylation increases moderately tension and Ca
bound to the troponin–tropomyosin complex. Regulatory myosin light chains modulate the rate and magnitude of contractility, as MyLC2 phosphorylation increases moderately tension and Ca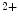 sensitivity at submaximal levels of Ca
sensitivity at submaximal levels of Ca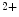 activation.
activation.
bound to the troponin–tropomyosin complex. Regulatory myosin light chains modulate the rate and magnitude of contractility, as MyLC2 phosphorylation increases moderately tension and Ca sensitivity at submaximal levels of Ca activation.Cardiac wall torsion and compression during systole enable a more efficient blood ejection from the ventricle. Torsion depends partly on the helical orientation of the cardiomyocytes. In the epicardial layer of the ventricular myocardium, the myofibers spiral in a left-handed helix from the apex to the base of the heart (the thumb denoting the upward spiral; Vol. 5, Chap. 5. Cardiomyocytes). In the endocardial layer of the ventricular myocardium, the myofibers have a right-handed helical orientation.
A spatial gradient of myosin light chain phosphorylation in the myocardium from high epicardial to low endocardial levels facilitates torsion by changing tension production and stretch activation response [323]. This spatial gradient of MyLC2 phosphorylation increases tension and decreases the stretch activation response of epicardial myofibers and conversely in the endocardial layer. Cardiac hypertrophy can be linked to a gain-in-function mutation.
The cardiac isoform of myosin-binding protein-C (cMyBPc) is an accessory protein of sarcomeric thick myosin filaments and a modulator of cardiomyocyte contraction in response to cardiac stimulation. Mutations in the MYBPC gene generate cardiac disease. The three isoforms of human MyBPc (fast and slow skeletal and cardiac) are encoded by three genes (MYBPC1–MYBPC3).
The cMyBPc molecule localizes to the central region of the A band, the C zone (Fig. 4.1). It is contained in 7–9 bands of 43-nm spacing among the 11 structurally regular transverse C-zone stripes [324]. These bands comprise three layers of myosin head crowns. Layer 1 is the very dense MyBPc layer, in which the C-termini of three MyBPc molecules wrap around the myosin filament, and layers 2 and 3 are simple myosin head crowns.
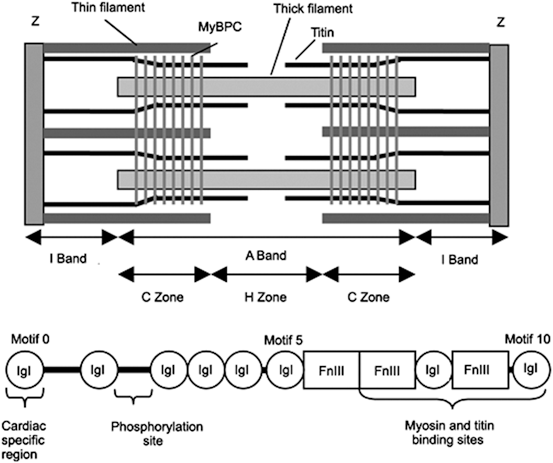
Fig. 4.1
Sarcomere striated configuration with lines, bands, and zones (Source: [324]). Position of MyBPc in the stretched sarcomere (top). MyBPc lodges in transverse stripes 43-nm apart in the C zone of the sarcomeric A band, where the thick myosin and thin actin filaments interact. Structure of the cardiac MyBPc isoform (bottom). The skeletal isoform of myosin-binding protein-C contains seven immunoglobulin-like and three fibronectin-3-like domains (C1–C10), a MyBPc-specific motif (M domain) between C1 and C2, and a Pro–Ala-rich sequence at the N terminus. The cardiac isoform has an additional N-terminal Ig-like domain (C0), 4 phosphorylation sites in the M domain (between motifs 1 and 2), and a 28-residue insert in the C5 domain. The cMyBPc protein tethers to the myosin filament in the C zone via its C-terminus (C8–C10 domains), whereas its N-terminus possesses binding sites for the myosin S2 motif and the myosin regulatory light chain. Motifs 7 to 10 link to myosin and titin
It is involved in sarcomere assembly, as it promotes polymerization of thick filaments, the C-termini binding to specific sites on titin and light meromyosin [324].
The extent of cMyBPc phosphorylation that occurs in response to adrenergic stimulation is correlated with increased cardiac contractility. Serine phosphorylation in M-domain cMyBPc by various kinases reduces the affinity of the N terminus for actin and myosin [325]. cMyBPc affects Ca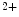 sensitivity similarly at long and short sarcomere lengths, as it reduces the probability of myosin binding to actin [326]. It also lowers the maximum shortening velocity of myofibers in the low-velocity phase (without influencing the high-velocity phase), as it contributes to an internal load that slows the maximum shortening velocity at low levels of activation once a given amount of active shortening has occurred [327]. Slowing of actin filament sliding is a mechanism used by cMyBPc to modulate cardiac contractility.
sensitivity similarly at long and short sarcomere lengths, as it reduces the probability of myosin binding to actin [326]. It also lowers the maximum shortening velocity of myofibers in the low-velocity phase (without influencing the high-velocity phase), as it contributes to an internal load that slows the maximum shortening velocity at low levels of activation once a given amount of active shortening has occurred [327]. Slowing of actin filament sliding is a mechanism used by cMyBPc to modulate cardiac contractility.
sensitivity similarly at long and short sarcomere lengths, as it reduces the probability of myosin binding to actin [326]. It also lowers the maximum shortening velocity of myofibers in the low-velocity phase (without influencing the high-velocity phase), as it contributes to an internal load that slows the maximum shortening velocity at low levels of activation once a given amount of active shortening has occurred [327]. Slowing of actin filament sliding is a mechanism used by cMyBPc to modulate cardiac contractility.The cMyBPc protein binds to myosin via its C-terminus and actin via its N-terminus, thereby modulating thin actin filament motility. It connects to actin close to the low Ca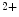 -binding site of tropomyosin, hence interfering with tropomyosin regulatory movements on actin. It indeed increases actin filament calcium sensitivity by binding to actin and displaces tropomyosin from its inhibitory position toward its high Ca
-binding site of tropomyosin, hence interfering with tropomyosin regulatory movements on actin. It indeed increases actin filament calcium sensitivity by binding to actin and displaces tropomyosin from its inhibitory position toward its high Ca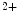 position to activate actin filament interaction with myosin, promoting filament sliding [325]. At high Ca
position to activate actin filament interaction with myosin, promoting filament sliding [325]. At high Ca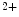 levels, cMyBPc has little effect on tropomyosin position and slows down actin filament sliding.
levels, cMyBPc has little effect on tropomyosin position and slows down actin filament sliding.
-binding site of tropomyosin, hence interfering with tropomyosin regulatory movements on actin. It indeed increases actin filament calcium sensitivity by binding to actin and displaces tropomyosin from its inhibitory position toward its high Ca position to activate actin filament interaction with myosin, promoting filament sliding [325]. At high Ca levels, cMyBPc has little effect on tropomyosin position and slows down actin filament sliding.The very long elastic molecule titin (or connectin) positions the myosin filaments at the center of a sarcomere, as it links them to the Z line.Their N- and C-termini localize to the Z and M line and respectively. The middle portion tethers to the myosin filament.
4.1.1.2 Costamere
The costamere3 is a structural and functional component of striated myocytes. This proteic complex consists of cytoskeletal proteins and signaling kinases. It couples the sarcomere to the sarcolemma. This subsarcolemmal proteic assembly encircles the myocyte and is circumferentially aligned with the Z disc, as it anchors myofibrils from the Z disc to the sarcolemma and extracellular matrix. Costameres are sites of force transmission [328].
The costamere is composed of the dystrophin-associated glycoprotein complex (DGC) and the integrin complex (Table 4.2). The dystrophin-associated glycoprotein complex is a structural unit and scaffold for signaling molecules at the sarcolemma. It is composed of dystroglycans and sarcoglycans as well as focal adhesion constituents. It maintains the structural integrity of myofibers by linking the extracellular matrix to the subsarcolemmal cytoskeleton. The costamere coordinates force transduction and intracellular signaling.
The dystrobrevin family comprises α- and β-dystrobrevin that are encoded by the DTNA and DTNB genes, respectively. The former is expressed predominantly in muscle and brain and the latter in nonmyocyte cells.
Table 4.2
Costamere constituents
Components | Constituents |
|---|---|
Structural components | Desmin, dysbindin, plectin, syncolin, |
β-synemin | |
Signaling components | α/β-syntrophin, α-catulin, calmodulin, |
myospryn, caveolin, NOS1, PKA, TRPC | |
Dystroglycan | (extracellular) α/β-dystroglycan, |
glycoprotein | (sarcolemmal) α–δ-sarcoglycan, sarcospan, |
complex | (subsarcolemmal) dystrophin, α-dystrobrevin |
Focal adhesion | Melusin, paxilin, |
(integrin complex) | talin, tensin, |
vinculin, zyxin, | |
ILK, LIMS, parvins | |
Matrix partners | Laminin, collagen-4 |
α-dystrobrevin is a component of the DGC that directly tethers to dystrophin. In striated myocytes, dystrobrevin and dystrophin localize to the cytoplasmic face of the sarcolemma. α-dystrobrevin also binds to intermediate filaments as well as syntrophin, a modular adaptor protein.4
α-syntrophin is the major isoform in skeletal and cardiac myocytes [329]. Syntrophin can coordinate the assembly of nitric oxide synthase NOS1, P38MAPKγ, transient receptor potential channels (TRPC), which modulates cation (calcium) entry, and calmodulin to the DGC.
α-dystrobrevin experiences alternative splicing that impacts its subcellular distribution and function in myocytes. Among the three major α-dystrobrevin isoforms, α-dystrobrevin-1 and α-dystrobrevin-2 localize to the sarcolemma. The former colocalizes with both dystrophin and utrophin and the latter only with dystrophin. α-dystrobrevin-1 to α-dystrobrevin-3 bind to the sarcoglycan complex [329]. α-dystrobrevin-1 and α-dystrobrevin-2 tether to dystrophin and syntrophin (but not α-dystrobrevin-3).
Additional α-dystrobrevin-binding partners include the constituents of the mechanical stabilizer intermediate filament syncoilin and β-synemin as well as dysbindin [329]. In striated myocytes, intermediate filaments encircle the Z disc, thereby connecting all adjacent myofibrils and linking the Z disc of the peripheral layer of cellular myofibrils to the sarcolemma. Syncoilin also binds to desmin, a muscle-specific intermediate filament protein. The α-dystrobrevin–syncoilin interaction provides another linkage between the DGC and cytoskeleton, which may be important for force transduction during contraction. β-synemin interacts with plectin, a linker protein of intermediate filaments to the Z disc. Dysbindin (or dystrobrevin-binding protein-1) is a ubiquitous sarcolemmal protein, the expression of which is relatively low in muscle [329]. It binds to α-dystrobrevin and myospryn.5 Myospryn may function as a docking platform for structural and signaling molecules, such as α-actinin-2 and protein kinase-A [329].
α-catulin is a ubiquitous binding partner of α-dystrobrevin-1 that localizes to nerve bundles and blood vessels. It may regulate α1d-adrenergic receptor signaling.
α-actinin binds to: (1) actin and the CRSP3–Tcap–titin complex6 to organize the local cell architecture and (2) to talin and vinculin to connect the sarcomeres to costameres and, hence, to integrins and the extracellular matrix constituents (e.g., collagen and laminin). The mechanosensor and mechanotransducer vinculin is also connected to intercellular junctions, not only integrin-based adhesion complexes, but also tight, adherens, and gap junctions.
The cytoplasmic domain of β-integrin binds integrin-linked kinase (ILK) and melusin. The former has structural and functional roles at the costamere, as it recruits various adaptors (e.g., LIM and senescent cell antigen-like domain-containing proteins [LIMS], 7 parvins [α-parvin, or actopaxin, and β-parvin, or affixin], and paxillin), cytoskeletal, and signaling molecules. In particular, it links to various kinases and phosphatases (e.g., protein Ser/Thr ILK-associated phosphatase [ILKAP], an inhibitor of ILK of the PPM1 category, 3-phosphoinositide-dependent [PI3K-dependent] protein kinase PDK1, which phosphorylates PKB, and PKB) [330]. Therefore, ILK is connected to the actin cytoskeleton, focal adhesion complex, and growth factor receptors.
Morerover, it phosphorylates protein kinase-B and other signaling effectors to regulate the muscular response to stretch [330].
In addition to their structural role, integrins and their associated adaptor proteins recruit to the plasma membrane signaling molecules used for the survival and growth of cardiomyocytes (e.g., focal adhesion kinase, ILK, P21-activated kinase, proline-rich tyrosine kinase PYK2, PKC, and Src) [330].
Furthermore, the focal adhesion complex can be linked to growth factor receptors, thereby enabling crosstalk between mechanotransduction and growth factor-mediated hypertrophic signals.
Costameric and sarcomeric proteins, particularly those of the Z disc, are components of the cardiac stretch sensor.
The mechanical stretch response can be impaired, whereas hypertrophic signaling triggered by humoral factors such as endothelin-1 can remain intact.
4.1.1.3 Mechanosensory Cadherin–Catenin-Mediated Adhesions
The N-cadherin complex links adjacent cardiomyocytes via adherens junctions (with α-, β-, and δ1-catenins and related partners), as do mechanosensory integrins vie focal adhesion complexes (with numerous cytoskeletal adaptors [e.g., filamin, paxillin, talin, and vinculin] and signaling effectors [e.g., focal adhesion kinase and small RhoA GTPase]), is sensitive to force transmission. Forces acting on N-cadherin–mediated adhesions elicit structural and functional changes in cardiomyocytes.
α-catenin is a major component of the mechanosensory cadherin–catenin complex that connects to the cell actin cytoskeleton. It homodimerizes or heterodimerizes with β-catenin. It localizes to regions of highest mechanical stress on cardiomyocytes [331]. Localization of α-catenin depends on N-cadherin topography, and hence on the degree of internal stress at the intercellular junction, that is, on changes in contractility. The α-catenin–cadherin complex indeed serves as a mechanosensory regulator of the cardiomyocyte cytoskeletal structure.
Sarcomeric remodeling depends on whether adhesion and force transmission is mediated by cadherins or integrins. Inhibition of myosin bound by integrins to fibronectin disrupts myofibril organization. On the other hand, inhibition of myosin bound by N-cadherins disturbs to a lower extent myofibril arrangement. These two mechanosensory cell adhesion types provide independent mechanisms for regulating sarcomeric organization. Lower myosin activity is preferential for cadherin-dependent assembly and preservation of sarcomeres [331]. Cadherin- and integrin-based junctions can sustain stresses of similar magnitudes, but subsequent cytoskeletal remodeling differs due to the specific function of proteic components and their interaction in assembling and orienting actin bundles in response to mechanical stresses.
4.1.2 Gene Mutations
More than 900 mutations in genes expressed in the cardiomyocyte can cause cardiomyopathies [332]. More than 400 mutations in 13 sarcomeric proteins are linked to cardiomyopathies (Table 4.3). Mutations in genes encoding sarcomeric constituents (e.g., cardiac α-actin, myosin light chain, cardiac β-myosin heavy chain, cardiac myosin-binding protein-C, cardiac troponin-I and -T, titin, and tropomyosin) are usually inherited in an autosomal-dominant manner and are missense mutations. Mutations in both the essential and regulatory myosin light chains cause a rare form of midventricular hypertrophic cardiomyopathy.
Table 4.3
Hypertrophic cardiomyopathies, genetic causes, and cellular pathology (Source: [332]). The MYH6 and MYH7 genes encode cardiac α-myosin heavy chain-6 and cardiac β-myosin heavy chain-7, respectively. Gene mutations can be common to multiple forms of cardiomyopathies
Mutated gene | Pathology |
|---|---|
Cardiac α-actin (ACTC1 gene) | Interstitial fibrosis |
α-myosin heavy chain (MYH6 gene) | Cardiomyocyte hypertrophy |
β-myosin heavy chain (MYH7 gene) | LV wall thickening |
Myosin light chains | Inflammation |
Myosin-binding protein-C | Increased cardiac mass |
α-tropomyosin (TPM1 gene) | Myofibrillar disarrangement |
Troponin-C | |
Troponin-I | Enlarged nuclei |
Troponin-T | ↓ LV volume |
MLP (muscle LIM protein) | |
Telothonin | |
Titin | Ventricular wall stiffness |
Vinculin |
Mutations of several sarcomeric components are associated with dilated cardiomyopathy and skeletal muscle myopathy. Major forms of muscular dystrophies are caused by abnormalities of the dystrophin-associated glycoprotein complex. Mutations in the dystrophin gene generate Duchenne and Becker muscular dystrophy. Duchenne muscular dystrophy in skeletal muscles and heart is characterized by the absence or slight production of functional dystrophin (Table 4.4). In Becker muscular dystrophy, a partly functional dystrophin is synthesized.
Table 4.4
Muscular dystrophy-associated cardiomyopathy, genetic causes, and cellular pathology (Source: [332]). Gene mutations can be common to multiple forms of cardiomyopathies
Mutated gene | Pathology |
|---|---|
Dystrobrevin | LV noncompaction |
Dystroglycan | Apoptosis |
Dystrophin | LV hypertrophy or dilation |
Sarcoglycan | Fibrosis |
Mutations in the sarcoglycan genes cause several types of sarcoglycan-deficient limb-girdle muscular dystrophy (SDLGMD).
Mutations in several Z-disc proteins of the cardiomyocyte sarcomere, which lead to disruption and dysfunction of the contractile apparatus, cause cardiomyopathies [333].
Ischemic and nonischemic cardiomyopathies share expression of some genes compared with controls, but numerous genes are differently expressed with respect to cardiomyopathy types and controls [334]. Among extracellular stimuli (ions, hormones, and mechanical stress), pathological myocardial remodeling is mainly induced by neurohormonal factors (angiotensin-2, endothelin-1, and catecholamines) via the Gq–PLCβ axis that activates the IP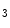 –Ca
–Ca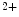 –PP3–NFAT and DAG–PKC pathways.
–PP3–NFAT and DAG–PKC pathways.
–Ca–PP3–NFAT and DAG–PKC pathways.On the other hand, adaptive cardiac (exercise-induced) hypertrophy that is particularly induced by growth hormone and insulin-like growth factor is associated with the PI3K–PKB–GSK3α/β pathway. Phosphoinositide 3-kinase-α downstream from the IGF1 receptor mediates adaptive cardiac growth (normal postnatal growth as well as growth in response to chronic exercise training) and protects the heart against pressure overload, dilated cardiomyopathy, and myocardial infarction.
4.2 Causes of Cardiomyopathies
Numerous inherited and acquired cardiomyopathies has been described, in addition to unclassified cardiomyopathies. They are categorized mainly into dilated, hypertrophic, and restrictive cardiomyopathies and arrhythmogenic right ventricular cardiomyopathy.
Many causes of cardiomyopathies exist (Table 4.5). Acquired cardiomyopathies can result from stress, diabetes, some chemotherapeutic agents, pregnancy, and alcohol intake. The most common cause is ischemic cardiomyopathy.
Table 4.5
Causes of cardiomyopathies
Cause | Disorder |
|---|---|
Cardiovascular | Ischemic heart disease, hypertension, |
valvular heart disease, cardiac amyloidosis, | |
tachycardia-induced cardiomyopathy, | |
idiopathic dilated cardiomyopathy, | |
idiopathic restrictive cardiomyopathy | |
Metabolic | Starvation, vitamin deficiency, glycogen storage disease, |
diabetes, hypo- and hyperthyroidism, acromegaly, | |
pheochromocytoma | |
Inflammatory, infectious | Secondary amyloidosis, sarcoidosis, |
Coxsackie-B viral infection, hepatitis C, | |
human immunodeficiency virus infection, | |
American trypanosomiasis (Chagas disease) | |
Toxic | Alcohol, cocaine, amphetamines, chemotherapy |
Genetic | Familial dilated cardiomyopathy, hemochromatosis, |
amyloidosis, noncompacted myocardium, | |
systolic dysfunction without dilation, | |
arrhythmogenic right ventricular cardiomyopathy | |
Pregnancy | Peripartum cardiomyopathy |
Cardiomyopathies are generally categorized into two groups: extrinsic and intrinsic cardiomyopathies.
1.
Extrinsic cardiomyopathies do not have myocardial origin, as they are associated with hypertension, ischemia (coronary heart disease), valvular dysfunction, metabolic and nutritional diseases, congenital heart disease, or diabetes.
2.
Intrinsic cardiomyopathies result from myocardial weakness without identifiable external cause. Intrinsic cardiomyopathy has many causes, such as drug and alcohol toxicity, myocarditis, infections, and genetic factors.
4.2.1 Specific Cardiomyopathies
Specific cardiomyopathies are referred to as myocardial diseases associated with specific cardiac or systemic disorders.
General system diseases,
such as connective tissue disorders (e.g., systemic lupus erythematosus, polyarteritis nodosa, rheumatoid arthritis, scleroderma, and dermatomyositis). They also include infiltrations and granulomas, such as sarcoidosis and leukemia.
Hypertensive cardiomyopathy
is a left ventricular hypertrophy associated with dilated or restrictive cardiomyopathy with features of cardiac failure.
Inflammatory cardiomyopathy
is defined by a myocarditis and cardiac dysfunction. It has idiopathic, autoimmune, and infectious (Trypanosoma cruzi, human immunodeficiency virus, enterovirus, adenovirus, and cytomegalovirus) forms.
Ischemic cardiomyopathy
is a dilated cardiomyopathy with impaired contractile performance not explained by the extent of coronary artery disease, that is, ischemic damage.
Metabolic cardiomyopathy
includes various categories:
Endocrine in the context of thyrotoxicosis, hypothyroidism, adrenal cortical insufficiency, pheochromocytoma, acromegaly, and diabetes mellitus;
Familial storage disease and infiltrations, such as hemochromatosis, glycogen storage disease, Hurler’s and Refsum’s syndromes, Niemann–Pick, Hand–Schüller–Christian, Fabry–Anderson, and Morquio–Ullrich diseases;
Deficiency and nutritional disorders, such as disturbances of potassium metabolism, magnesium deficiency, kwashiorkor, anemia, beri-beri, and selenium deficiency;
Cardiac amyloidoses that can be primary, secondary, familial, and hereditary, including familial Mediterranean fever and senile amyloidosis.
Muscular dystrophies
that comprise Duchenne, Becker-type, and myotonic dystrophies.
Neuromuscular disorders
that encompass Friedreich’s ataxia, Noonan’s syndrome, and lentiginosis.
Peripartal cardiomyopathy
that can first manifest in the peripartum period.
Sensitivity and toxic reactions
to alcohol, catecholamines, anthracyclines, irradiation, among others.
Valvular cardiomyopathy
is related to a ventricular dysfunction that is out of proportion to the abnormal loading conditions.
4.3 Classification of Cardiomyopathies
Intrinsic cardiomyopathies are usually classified into four main categories (Table 4.6;[210]):
1.
Dilated cardiomyopathy with left ventricle enlargment and reduced pumping function;
2.
Hypertrophic cardiomyopathy due to mutations in genes encoding sarcomeric proteins;
3.
Restrictive cardiomyopathy with stiff ventricular walls; and
4.
Arrhythmogenic right ventricular cardiomyopathy with fibrous tissue.
4.4 Mitochondria in Cardiomyopathies
Like in normal hearts, two populations of mitochondria exist in cardiomyopathies at least in hamsters [335]: (1) subsarcolemmal mitochondria beneath the sarcolemma and (2) interfibrillar mitochondria among the myofibrils.
Mitochondrial enzymes (enoylCoA hydratase, hydroxyacylCoA dehydrogenase, and citrate synthase) are depressed in cardiomyopathies. The content of coenzyme-A remains unaltered, but carnitine amount is reduced.
The two heart mitochondrial populations are distinctly affected, as they react differently in cardiomyopathy. Oxidative phosphorylation in subsarcolemmal mitochondria is normal, whereas it decays in interfibrillar mitochondria. The amount of mitochondrial respiratory supercomplexes mtRSC1 (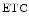 cI–
cI–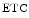 cIII–
cIII–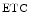 cIV) of the electron transport chain, the major form of the so-called respirasome, can decay, thereby reducing the efficiency of electron transfer and energy production.
cIV) of the electron transport chain, the major form of the so-called respirasome, can decay, thereby reducing the efficiency of electron transfer and energy production.
cI–cIII–cIV) of the electron transport chain, the major form of the so-called respirasome, can decay, thereby reducing the efficiency of electron transfer and energy production.Table 4.6
Classification of cardiomyopathies. (Source: [210])
Type | Features | Causes |
|---|---|---|
Dilated | Dilated | Ischemic, immune, toxic, |
Left or both ventricles | alcoholic, valvular, | |
Impaired contraction | idiopathic, genetic (familial) | |
Hypertrophic | L(R)V hypertrophy or both | Genetic |
Restrictive | Reduced diastolic filling | Amyloidosis, |
Normal or near-normal | endomyocardial fibrosis, | |
systolic function | idiopathic | |
ARVC | Fibrofatty replacement | Genetic, idiopathic, |
of RV myocardium | Naxos disease | |
Unclassified | Atypical | Fibroelastosis, |
noncompacted myocardium, | ||
systolic dysfunction with | ||
slight dilation, | ||
mitochondrial disease |
4.5 Altered Cardiomyocyte Morphology and Function
Compensatory responses, such as changes in gene expression and cellular morphology and metabolic shifts in cardiomyocytes, can initially maintain the cardiac function in cardiomyopathy.
4.5.1 Cardiomyocyte Hypertrophy
Cardiomyocyte hypertrophy (Sect. 3.2) aimed at normalizing increased parietal stress increases the wall thickness, but decreases inner dimensions of the ventricular chambers. Whereas the initial phase of hypertrophy can be compensatory, persistent adverse hypertrophy leads to heart failure.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


