1995 (WHO)
2006 (AHA)
2008 (ESC)
Idiopathic
Primary
Familial/genetic
DCM
Genetic
or
HCM
HCM
Non–familial
RCM
ACM
ARVC
LVNC
HCM
Unclassified
Glycogen storage defects
DCM
Conduction defects
RCM
Specific
Mitochondrial myopathies
ARVC
Ion channel disorders
Unclassified
Mixed:
DCM
RCM
Acquired:
Inflammatory (myocarditis)
Stress-provoked (Tako–Tsubo)
Peripartum
Tachycardia-induced
Infants of insulin-dependent diabetic mothers
Secondary
Finally, the ESC classification is based on the main morphological patterns but then makes distinctions between familial and non-familial entities. This classification does not include channelopathies as a type of cardiomyopathy.
Remarks |
Hypertrophic and arrhythmogenic cardiomyopathies are the most common cause of sudden death associated with sports, among the young |
More than one half of heart transplants are performed on patients affected by a cardiomyopathy |
8.2 Hypertrophic Cardiomyopathy
Hypertrophic cardiomyopathy (HCM) is a heart muscle disease characterised by the presence of significant primary hypertrophy in the left ventricle (in adults ≥ 15 mm), which means that it is not caused by disorders leading to increased LV afterload (resistance to blood ejection). In clinical practice, conditions than can provoke compensatory or secondary ventricular hypertrophy are mainly hypertension and aortic stenosis.
HCM is a relatively common clinical entity with an incidence of 1 in every 500 individuals. Familial origin occurs in 50–60 % of the cases, presenting an autosomal dominant pattern of transmission in most of the cases, with incomplete penetrance.
Mutations in several genes have been identified as associated with HCM. The majority of the mutated genes encode sarcomeric proteins, which are proteins that form the contractile apparatus of the myocyte.
Clinically, it is important to highlight the great variability in symptomatology among these patients, not only in clinical presentation but also evolution and prognosis. Patients can report chest pain, dyspnoea on exertion, dizziness or syncope. The ECG usually shows a pattern of LV hypertrophy with increased voltage in the left precordial leads, I and aVL, and prominent and pathological Q waves, although deep and narrow (“dagger-like”). Alterations in the T waves and depression in the ST segment are also common. A definitive diagnosis is carried out with echocardiography by measuring the parietal thickness at different planes and projections, and discarding other clinical entities that could cause a similar degree of hypertrophy. In adults, HCM is diagnosed when the maximal wall thickness is greater or equal to 15 mm measured in end-diastole.
A small subgroup of patients have a particularly malignant course with episodes of sudden cardiac death (SCD) due to ventricular arrhythmias. In order to identify patients at higher risk, who would benefit from an automatic implantable cardiac defibrillator (ICD), a prognostic stratification should be done taking into account several risk factors associated with SD.
HCM can be considered obstructive or non-obstructive depending on the presence of obstruction of the left ventricular outflow tract (LVOTO). One third of patients with HCM have a significant obstruction (>30 mmHg) at rest and an additional third develops obstruction on exertion but not at rest. Based on the location at which hypertrophy is more manifest, HCM can be clinically divided in concentric, asymmetric with sigmoid septal contour (septal HCM), asymmetric with reverse pattern and apical forms. Gross and microscopic features are shown in Table 8.2.
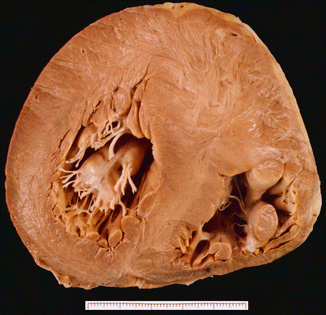
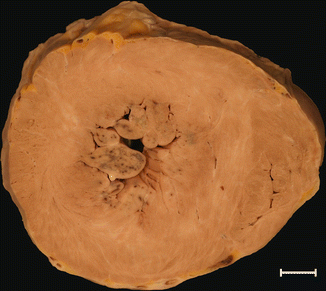
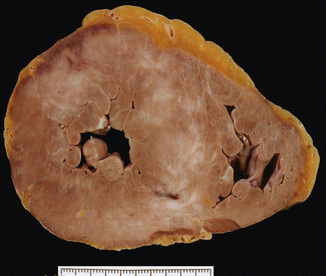
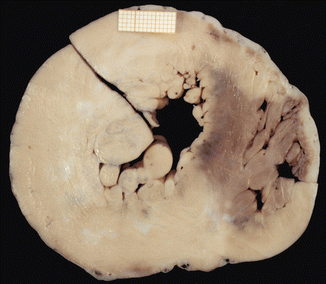
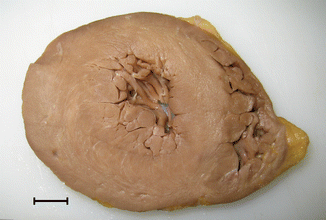
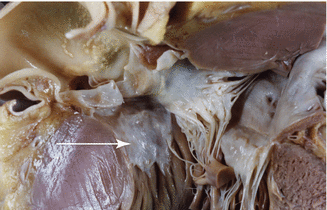
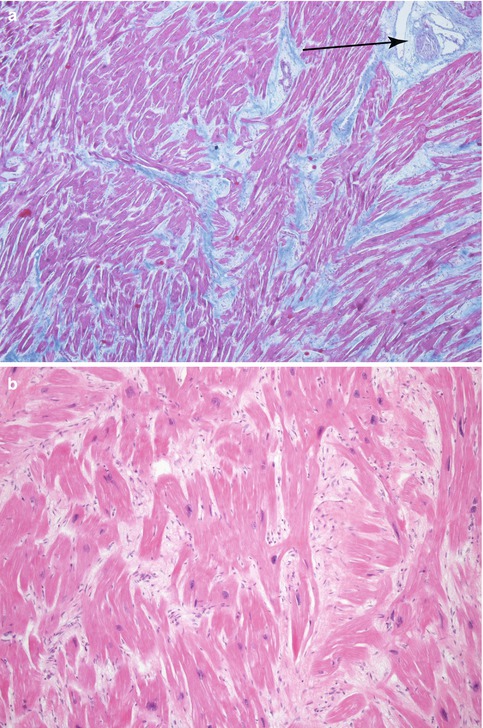
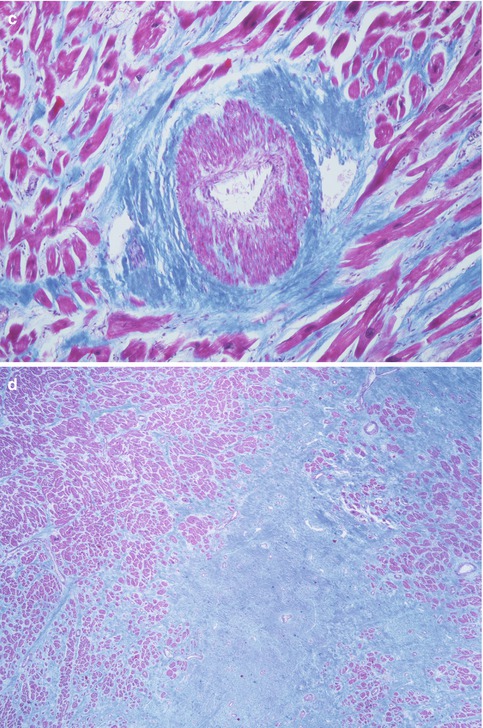
Table 8.2
Pathology of HCM
Macroscopic features: |
Myocardial hypertrophy (heart weight can exceed 500 g) |
Predominance of left ventricular hypertrophy, although the right ventricle may also be involved |
Based on the location, LV hypertrophy adopts several forms: |
Type I: hypertrophy of the anterior part of the ventricular septum, usually basal (Fig. 8.1) |
Type II: hypertrophy affecting the entire septum, without affecting the free wall of the LV (Fig. 8.2) |
Type III: hypertrophy of the septum and the anterior wall of the LV (Fig. 8.3) |
Prominent trabeculae and papillary muscles |
“Whorled” appearance of the myocardium |
White scarring foci randomly distributed (do not follow the vessels as in scarring in infarcts) |
Plaque of subaortic endocardial fibrosis due to the contact of the anterior mitral valve leaflet (it is not pathognomonic, it also may appear in aortic valve stenosis) (Fig. 8.6) |
LV dilatation may be present at end-stages of the disease |
Microscopic features: (Fig. 8.7) |
Myocyte hypertrophy, with hyperchromatic nuclei |
Myocyte disarray, with myocytes arranged at angles or adopting a whirling appearance or as fascicles organised perpendicularly |
Myofibril disarray |
Increased interstitial cellularity |
Thickened intramyocardial coronary arteries due to hypertrophy, disarray in the medial layer and intimal fibrosis, which contributes to ischaemia and myocardial fibrosis |
Evolution towards interstitial or replacement fibrosis |
Foci of myocarditis in some cases |
Fig. 8.1
HCM type I. Sudden death in a 22-year-old male. Heart weight was 505 g. Asymmetric HCM with major hypertrophy of the anterior portion of the ventricular septum (4 cm thickness) and whirling appearance of the myocardium
Fig. 8.2
HCM type II. A 27-year-old woman who died suddenly at home. Severe HCM with predominant involvement of the septum. Microinfarctions in papillary muscles and trabeculae can be observed, caused by small vessel disease (viewed microscopically)
Fig. 8.3
HCM type III. A 24-year-old male who was diagnosed with obstructive HCM at the age of 8. He died suddenly at work while getting changed. Heart weight was 602 g and showed major hypertrophy of the ventricular septum and the anterior wall of the LV with extensive foci of fibrosis. Microscopic study revealed intense dysplasia in the intramural coronary arteries (Image reproduced with permission of Aguilera and Suárez-Mier 2003)
Fig. 8.4
HCM type IV. A 13-year-old boy who died suddenly at school during physical education class. Heart weight was 344 g and gross examination showed hypertrophy of the free wall of the LV (15 mm) with multiple scarring foci. Septum thickness was preserved (11 mm)
Fig. 8.5
Apical HCM. A 60-year-old male. Sports-related sudden death. Heart weight: 521 g. LV: 1.8 cm in posterior and lateral walls and 1.3 cm in anterior wall and septum, RV: 0.5 cm. Apical and posterolateral LV hypertrophy. 1 cm
Fig. 8.6
Subaortic endocardial fibrosis. A 36-year-old male, diagnosed with moderate obstructive HCM. Subaortic endocardial fibrosis. Specular image of the anterior leaflet of the mitral valve (arrow)
Fig. 8.7
Microscopic features of HCM. (a) Myocardium with architectural disarray, fibrosis and thickened intramural arteries (arrow). (b) Hypertrophic myocytes perpendicularly distributed and with hyperchromatic nuclei. (c) Intramural coronary artery dysplasia with thickened wall and medial layer alterations. (d) Patch of fibrosis. (a, c, d: Masson’s trichrome)
8.2.1 Idiopathic Left Ventricular Hypertrophy
Idiopathic left ventricular hypertrophy is characterised by concentric LV hypertrophy in the absence of hypertension, valve disease or aortic disease. Microscopically, myocyte disarray, characteristic of HCM, is not found (Fig. 8.8). Clinically, it is not a recognised entity and it is included within HCM. So far, limited molecular studies have been carried out in these patients and results are diverse: in some cases, mutations in genes encoding sarcomeric proteins (as in HCM) have been described, but in other cases no mutation was found. This form of LV hypertrophy has been associated with sports-related SCD and, from a pathologist’s point of view, a differential diagnosis with the athletes’ heart must be done.
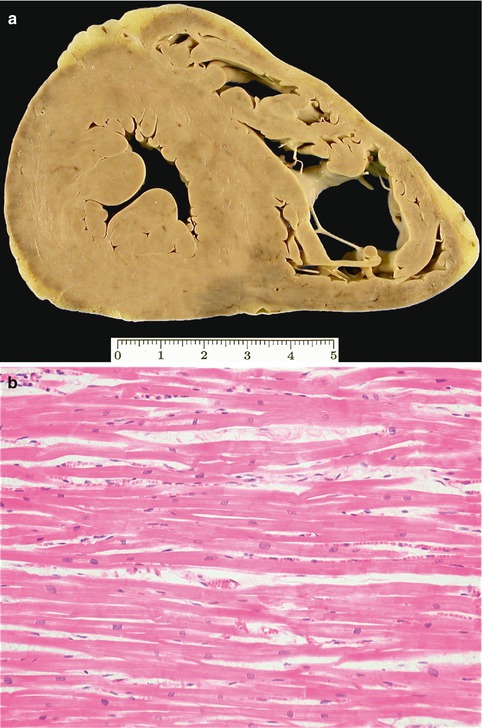
Fig. 8.8
Idiopathic left ventricular hypertrophy. A 15-year-old teenager suffered sudden death after cycling for 30 km. (a) Heart weight of 490 g and thickness of the LV walls of 15 mm. (b) Myocytes are hypertrophic and neatly arrayed. Cardiac family screening was negative
8.3 Arrhythmogenic Cardiomyopathy
Arrhythmogenic cardiomyopathy (ACM) is a condition characterised by fibrous or fibro-fatty replacement of the myocardium. ACM was initially described as arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D), but during the last decade it has been demonstrated that the disease can affect both ventricles or even exclusively or predominantly the LV. A high density of ventricular arrhythmias, with varying degrees of complexity, is frequently found in patients with ACM. Characteristic features of ACM include the presence of ventricular wall thinning, associated with aneurysms preferentially located at the infundibular, subtricuspid or apical levels in the RV, or regional wall motion abnormalities in the LV.
A family history of the disease is present in approximately 50 % of patients, and inheritance shows an autosomal dominant pattern with variable expressivity and incomplete penetrance. Mutations in genes encoding desmosomal proteins constitute the most commonly found genetic defect in these patients. Desmosomes are molecular protein complexes that participate in the maintenance of cellular structure and cell-to-cell adhesions. Defects in these proteins can lead to separation between cells due to defective intercellular junctions, which can cause apoptosis and death of myocytes, and ultimately their replacement with fibrous and adipose tissue.
ACM usually debuts in adolescents and before the fourth decade of life. Clinical presentation can be very variable, from progressive heart failure to a mainly arrhythmogenic disease with cardiac arrest as the first manifestation. Ventricular arrhythmias are very common, hence the name of the disease, and can vary from isolated ventricular extrasystoles to sustained ventricular tachycardia (VT) or ventricular fibrillation (VF).
ACM must be suspected in individuals suffering frequent and more or less complex ventricular arrhythmias, in patients that have had a cardiac arrest, and in those with RV dilatation non-attributab. to pulmonary disease or to a systemic-pulmonary shunt. Diagnosis may be difficult, and in the clinical practice ACM is diagnosed when the patient fulfils several major and/or minor criteria. These criteria are established depending on the degree of sensitivity, specificity and predictive value of the regional wall motion abnormalities identified through imaging techniques such as echocardiography or MRI, through ECG abnormalities, ventricular arrhythmias, familial background/genetic, and histopathological studies.
Treatment of ACM is focused on the prevention of SCD and the management of heart failure. Competitive physical exercise is contraindicated. Pharmacological treatment is initiated in patients with ventricular arrhythmias or progressive ventricular dysfunction. An implantable cardiac defibrillator could be an option for patients that have suffered a cardiac arrest, syncope, or badly-tolerated ventricular arrhythmias, and in patients with severe left or right ventricular dysfunction, as a bridge to heart transplantation. Radiofrequency ablation may diminish the occurrence and complexity of the arrhythmias suffered by these patients, although this is a transient solution.
Three forms of ACM exist depending on the ventricle involved:
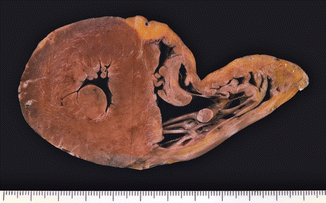
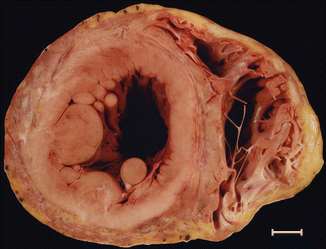
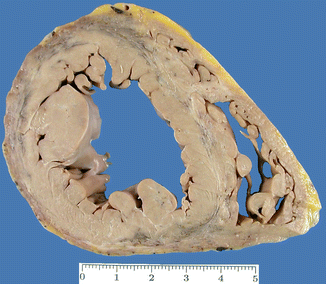
Fig. 8.9
Arrhythmogenic right ventricular cardiomyopathy (ARVC). A 29-year-old male who died suddenly while playing tennis. The RV is dilated with a thinned and yellow wall. No alterations were found in the LV
Fig. 8.10
Arrhythmogenic biventricular cardiomyopathy. A 23-year-old male who died while out having a drink (alcohol). Heart weight: 483 g. Involvement of the subepicardial half of the myocardium, the free wall of the LV, and the middle zone of the ventricular septum, adopting a circumferential pattern. The RV is dilated, with only the trabecular myocardium preserved (Image reproduced with permission of Aguilera and Suárez-Mier 2003)
Fig. 8.11
Arrhythmogenic left ventricular cardiomyopathy (ALVC). A 37-year-old woman, 12 weeks pregnant, who died suddenly while watching TV. Family history of premature SD of her father and two of her sisters (no autopsy was performed). ACM with left predominance and with a circumferential pattern. The affected myocardium appears darker and depressed due to the fixative solution
The pathologic characteristics of ACM are shown in Table 8.3.
Table 8.3
Pathology of ACM
Macroscopic features: |
Increased heart weight |
Fibrous or fibro-fatty replacement of myocardial tissue that initiates in the subepicardial ventricular region (in the RV only the trabecular myocardium is preserved) |
Patchy or diffuse myocardial lesion |
Circumferential involvement of the LV with an impaired ventricular septum |
Ventricular dilatation with formation of aneurysms, especially in the RV |
Microscopic features: |
Alterations in myocyte morphology with apoptosis and cytoplasmic vacuolation (Fig. 8.14) |
Frequent foci of inflammatory cell infiltrates < div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|